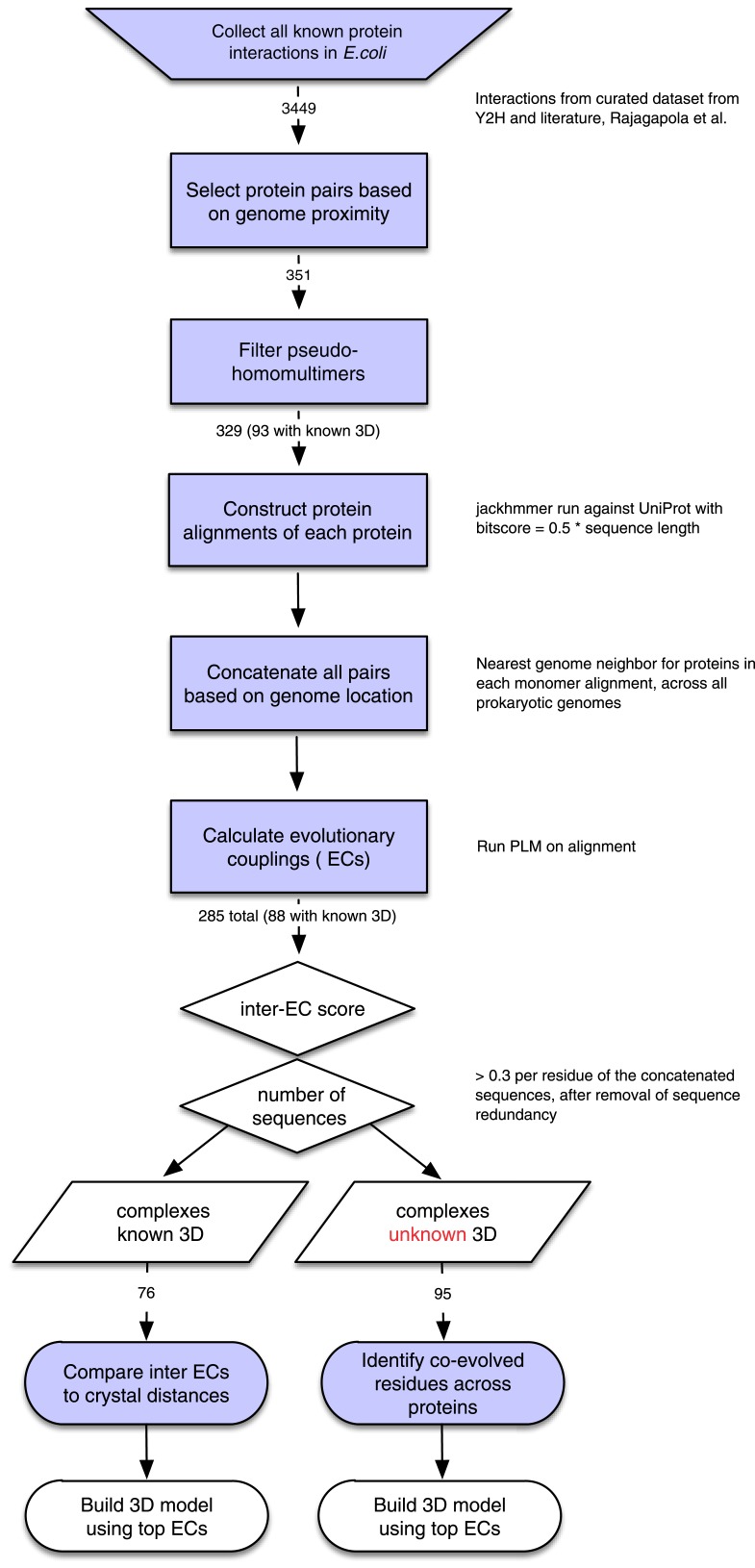
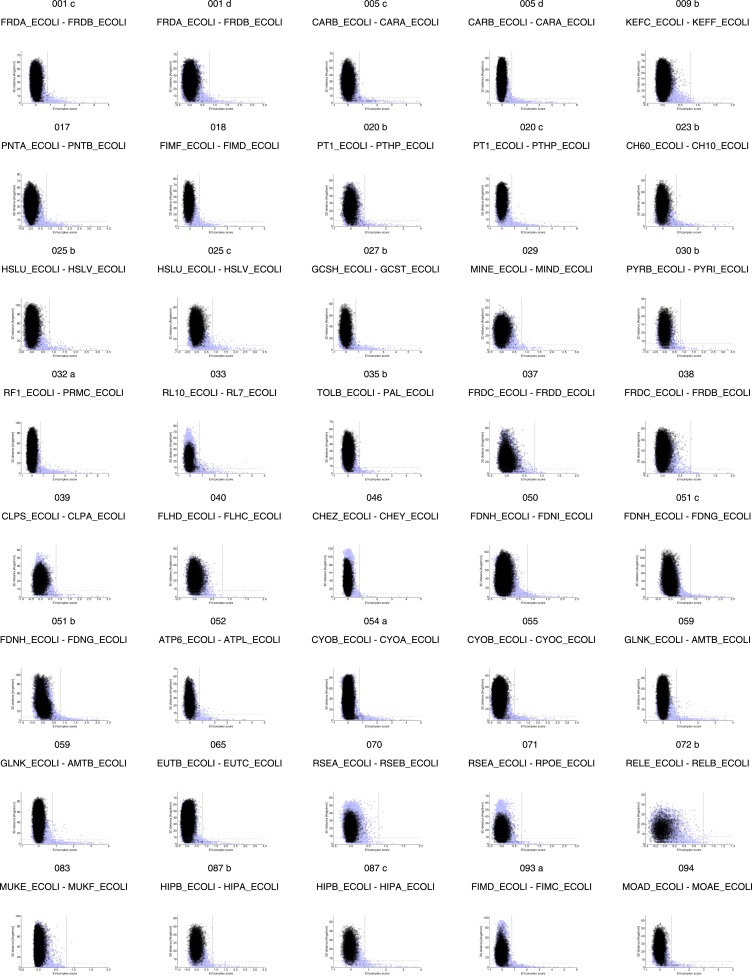
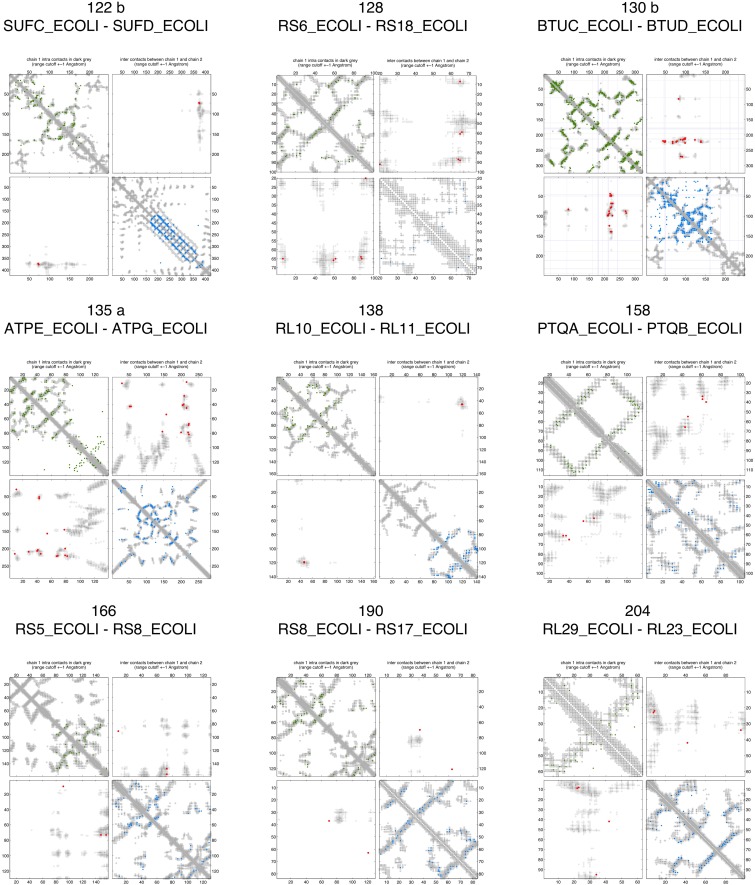
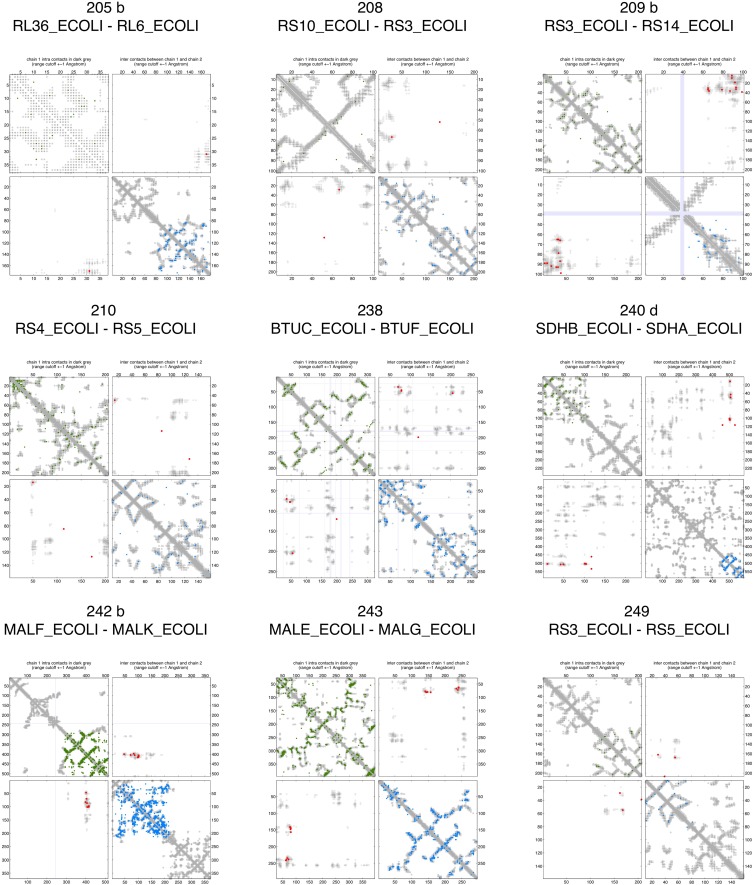
Sequence co-evolution gives 3D contacts and structures of protein complexes
- PMID: 25255213
- PMCID: PMC4360534
- DOI: 10.7554/eLife.03430
Sequence co-evolution gives 3D contacts and structures of protein complexes
Abstract
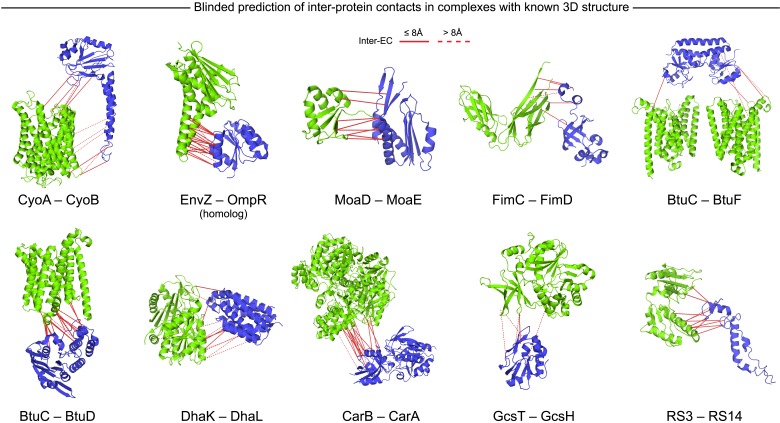
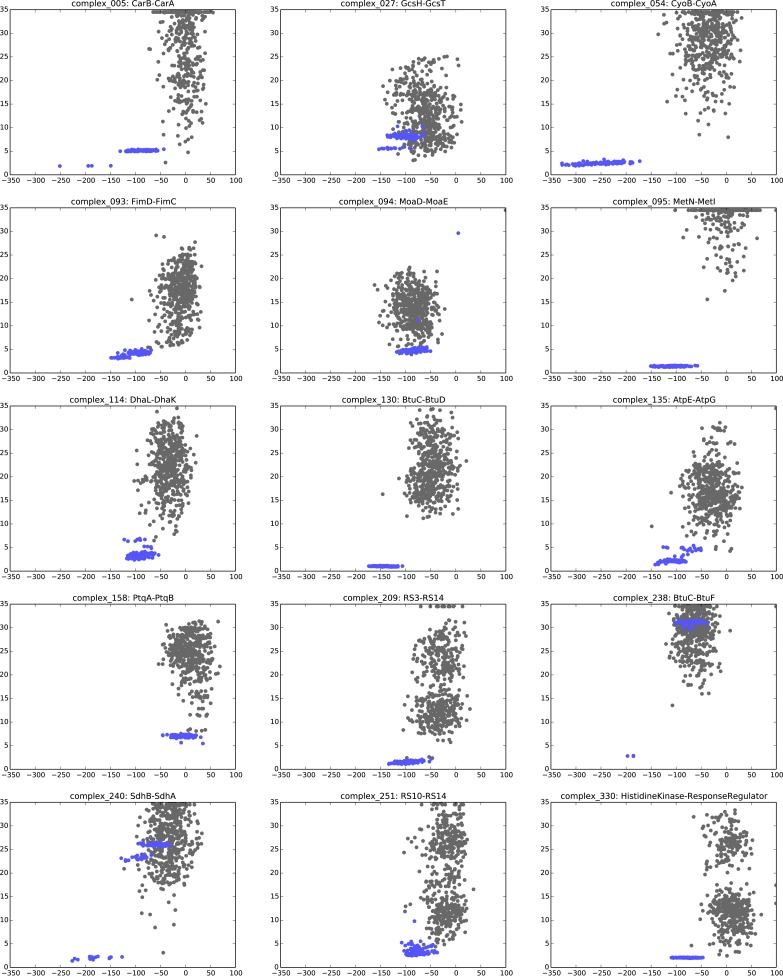
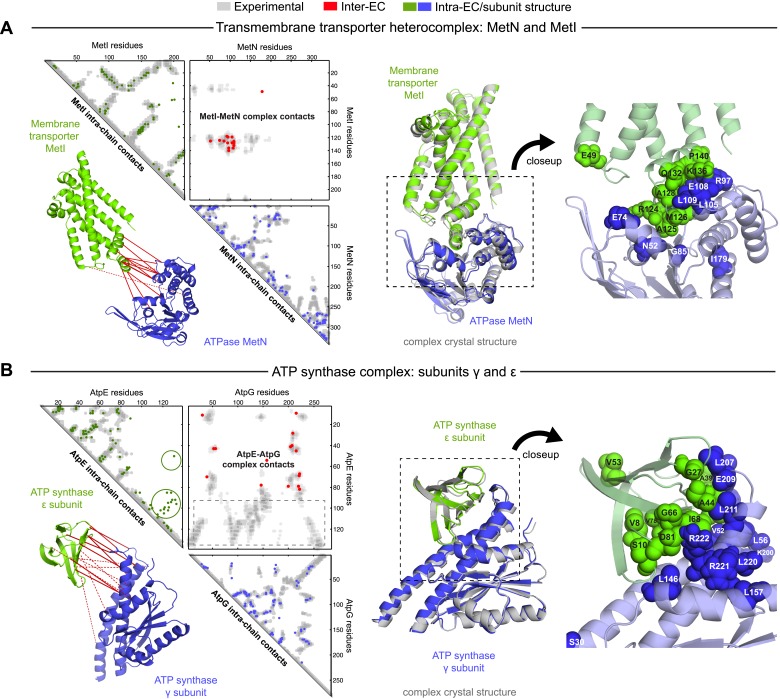
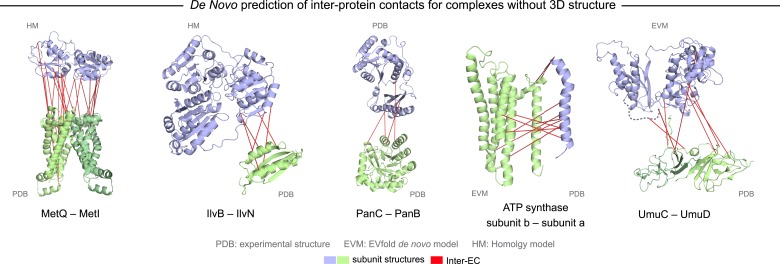
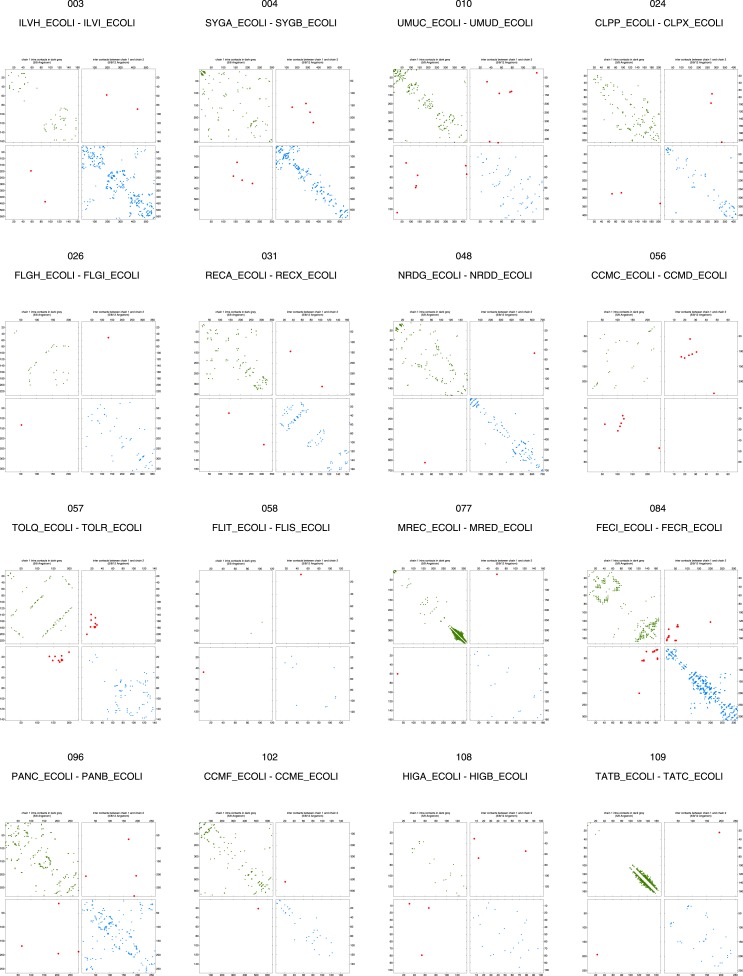
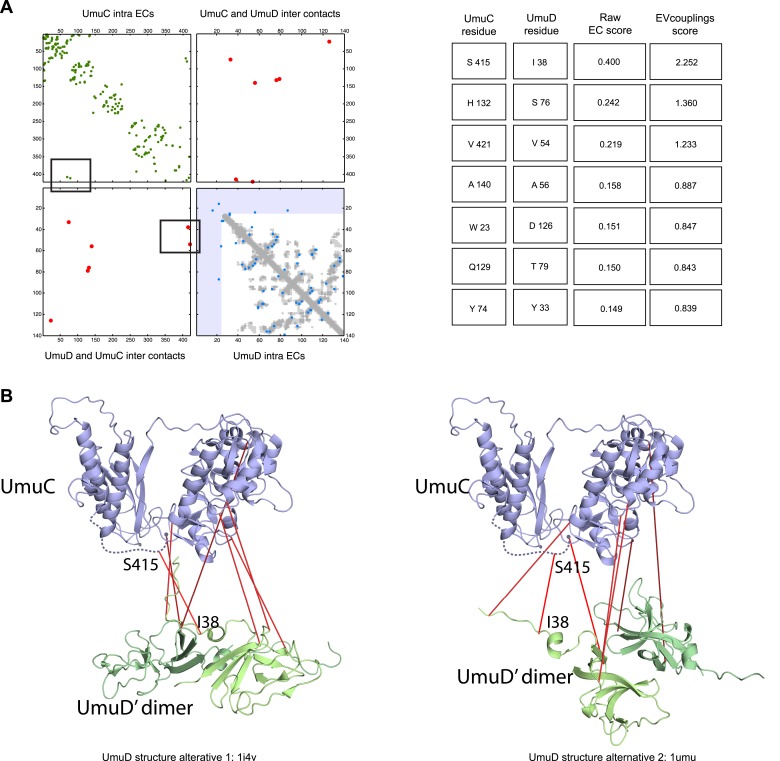
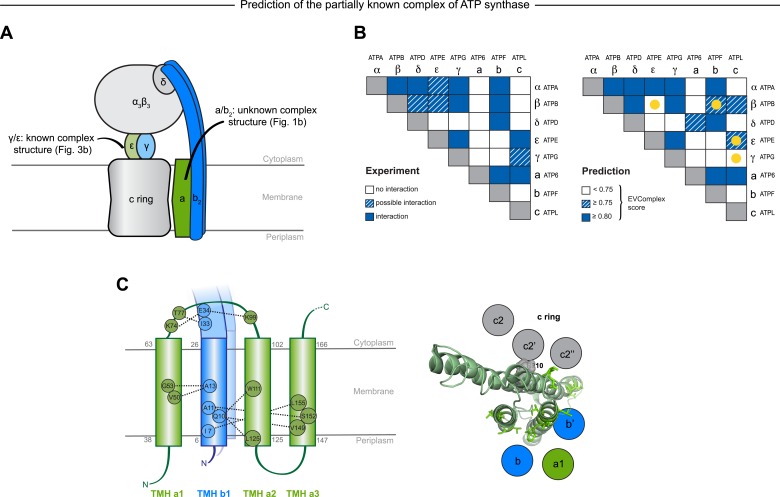
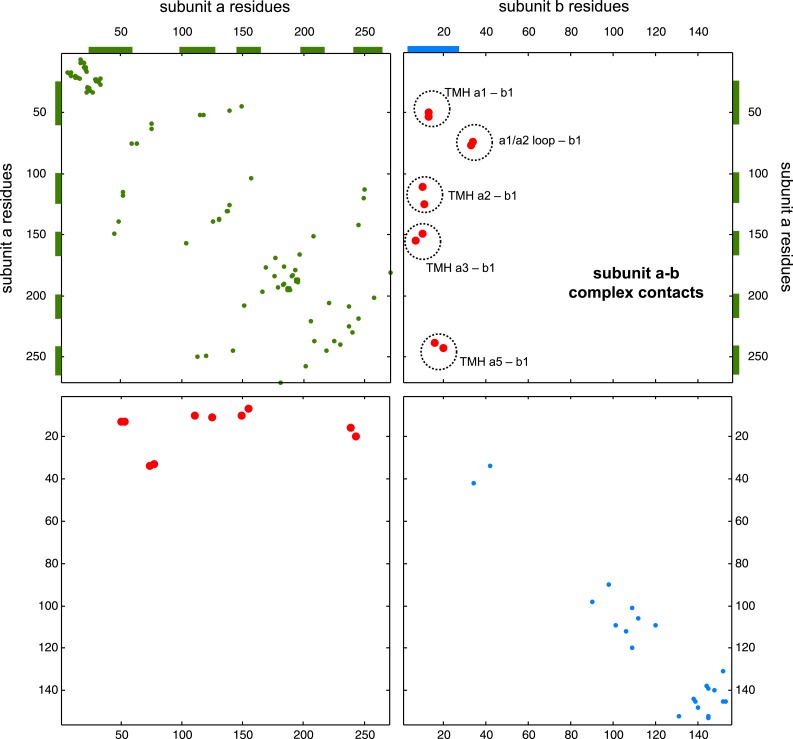
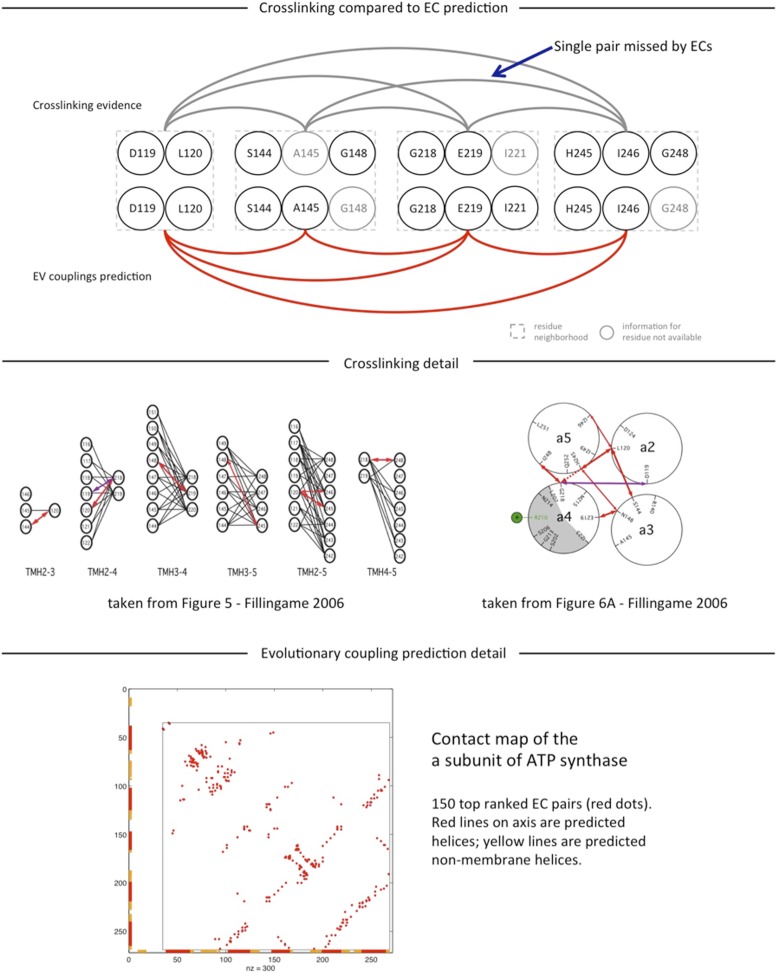
Protein-protein interactions are fundamental to many biological processes. Experimental screens have identified tens of thousands of interactions, and structural biology has provided detailed functional insight for select 3D protein complexes. An alternative rich source of information about protein interactions is the evolutionary sequence record. Building on earlier work, we show that analysis of correlated evolutionary sequence changes across proteins identifies residues that are close in space with sufficient accuracy to determine the three-dimensional structure of the protein complexes. We evaluate prediction performance in blinded tests on 76 complexes of known 3D structure, predict protein-protein contacts in 32 complexes of unknown structure, and demonstrate how evolutionary couplings can be used to distinguish between interacting and non-interacting protein pairs in a large complex. With the current growth of sequences, we expect that the method can be generalized to genome-wide elucidation of protein-protein interaction networks and used for interaction predictions at residue resolution.
Keywords: E. coli; co-evolution; evolutionary biology; genomics; interactions; protein.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures