

All-atom crystal simulations of DNA and RNA duplexes
- PMID: 25255706
- PMCID: PMC4339523
- DOI: 10.1016/j.bbagen.2014.09.018
All-atom crystal simulations of DNA and RNA duplexes
Abstract
Background: Molecular dynamics simulations can complement experimental measures of structure and dynamics of biomolecules. The quality of such simulations can be tested by comparisons to models refined against experimental crystallographic data.
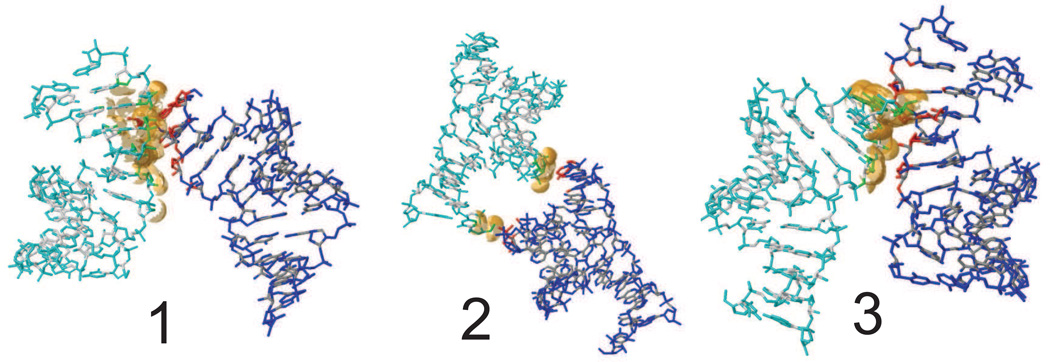
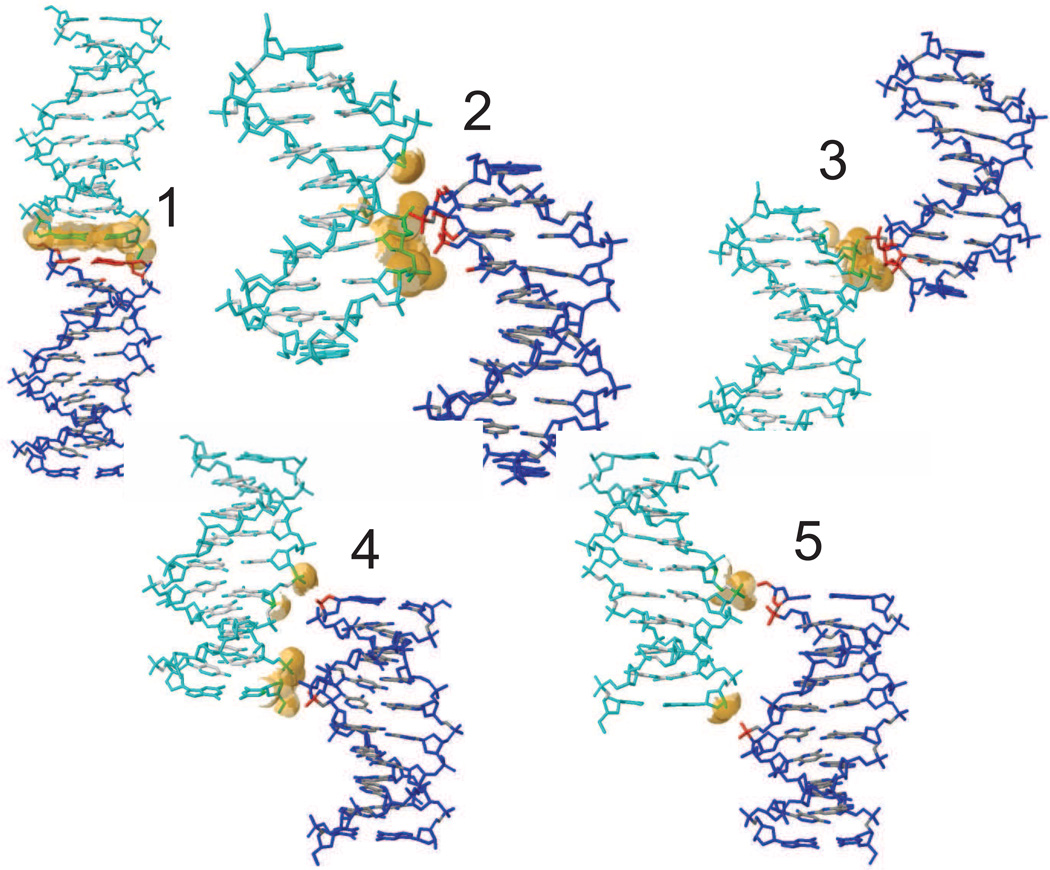
Methods: We report simulations of DNA and RNA duplexes in their crystalline environment. The calculations mimic the conditions for PDB entries 1D23 [d(CGATCGATCG)2] and 1RNA [(UUAUAUAUAUAUAA)2], and contain 8 unit cells, each with 4 copies of the Watson-Crick duplex; this yields in aggregate 64μs of duplex sampling for DNA and 16μs for RNA.
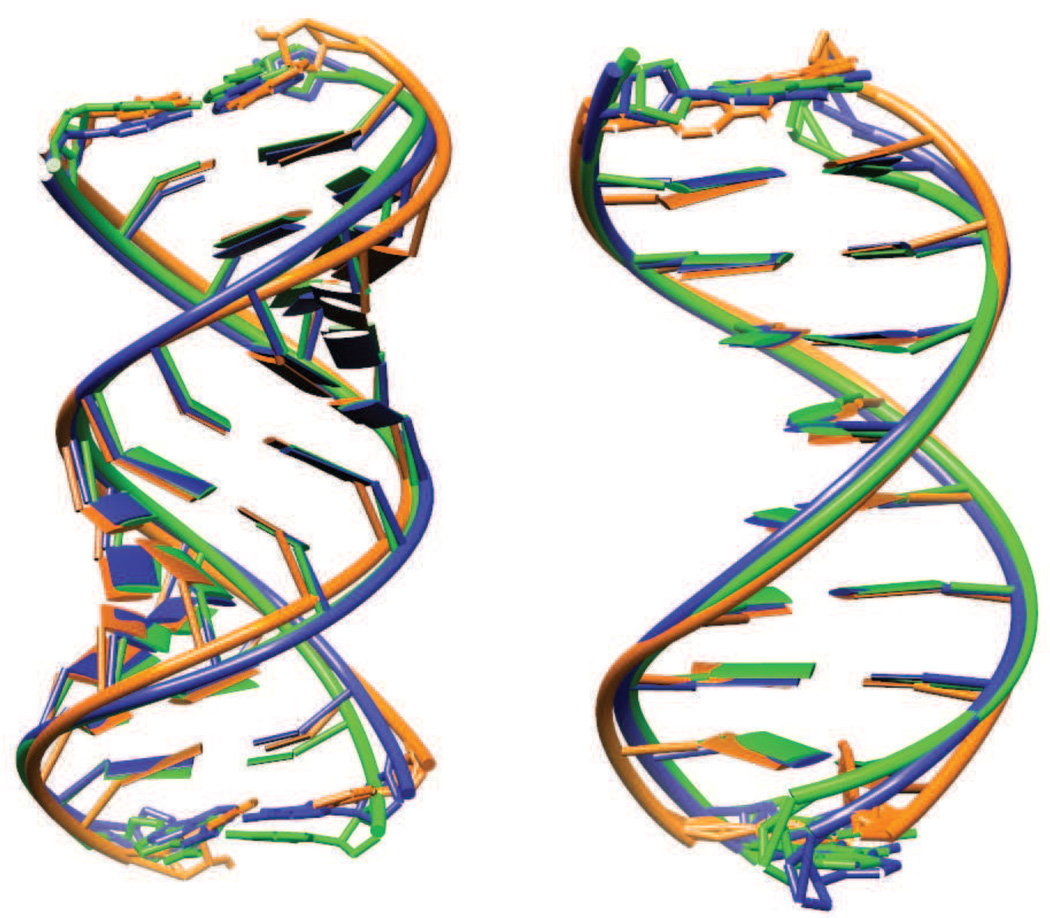
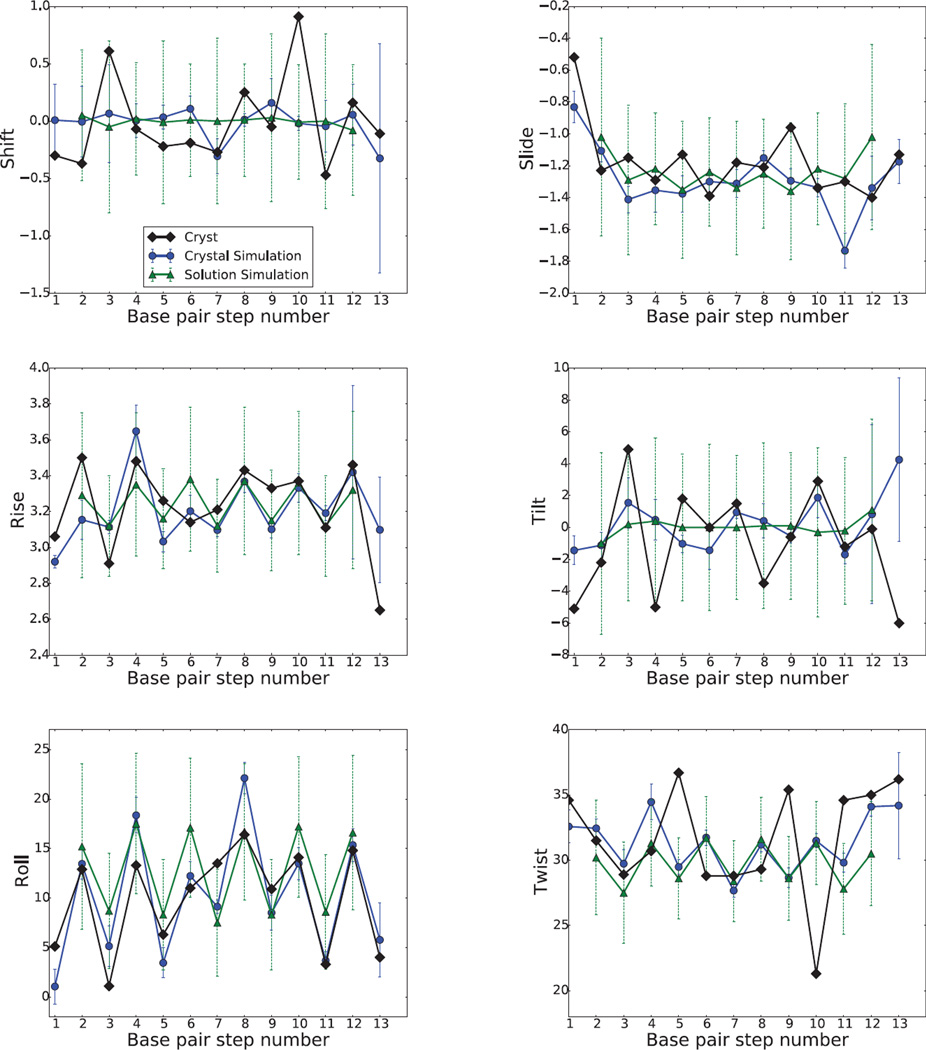
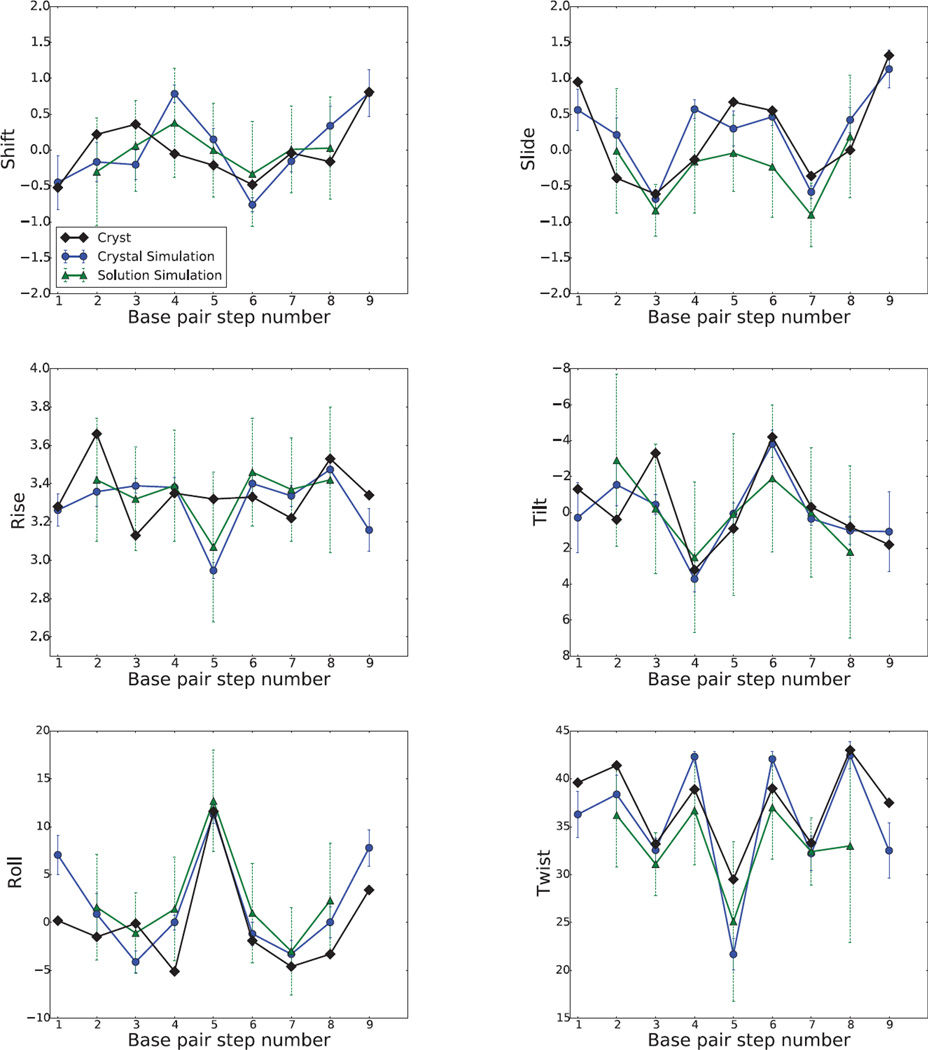
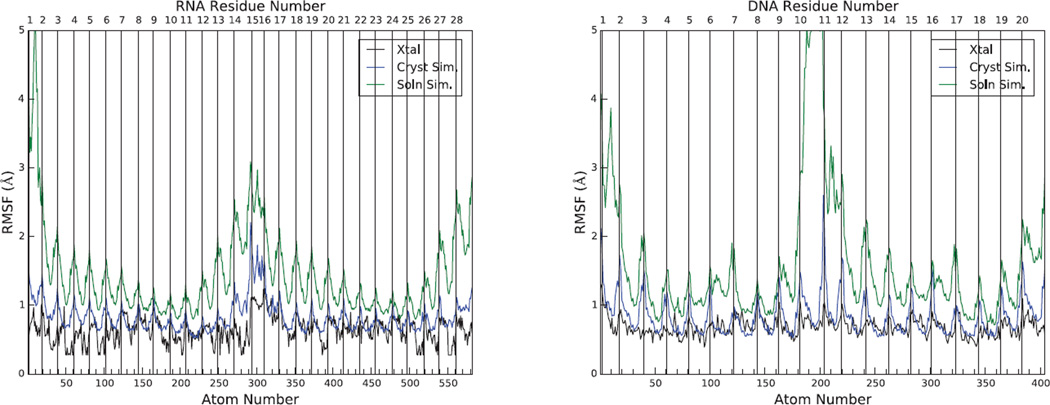
Results: The duplex structures conform much more closely to the average structure seen in the crystal than do structures extracted from a solution simulation with the same force field. Sequence-dependent variations in helical parameters, and in groove widths, are largely maintained in the crystal structure, but are smoothed out in solution. However, the integrity of the crystal lattice is slowly degraded in both simulations, with the result that the interfaces between chains become heterogeneous. This problem is more severe for the DNA crystal, which has fewer inter-chain hydrogen bond contacts than does the RNA crystal.
Conclusions: Crystal simulations using current force fields reproduce many features of observed crystal structures, but suffer from a gradual degradation of the integrity of the crystal lattice.
General significance: The results offer insights into force-field simulations that test their ability to preserve weak interactions between chains, which will be of importance also in non-crystalline applications that involve binding and recognition. This article is part of a Special Issue entitled Recent developments of molecular dynamics.
Keywords: Crystal; Molecular dynamics; Nucleic acids.
Copyright © 2014 Elsevier B.V. All rights reserved.
Figures









References
-
- Kim HS, Mhin BJ, Yoon CW, Wang CX, Kim KS. A theoretical study of a Z-DNA crystal: Structure of counterions, water and DNA molecules. Bull. Korean Chem. Soc. 1991;12:214–219.
-
- MacKerell AD, Jr, Wiórkiewicz-Kuczera J, Karplus M. An all-atom empirical energy function for the simulation of nucleic acids. J. Am. Chem. Soc. 1995;117:11946–11975.
-
- Cheatham TE, III, Miller JL, Fox T, Darden TA, Kollman PA. Molecular dynamics simulations on solvated biomolecular systems: The particle mesh Ewald method leads to stable trajectories of DNA, RNA and proteins. J. Am. Chem. Soc. 1995;117:4193–4194.
-
- York DM, Yang W, Lee H, Darden T, Pedersen LG. Toward the accurate modelling of DNA: The importance of long-range electrostatics. J. Am. Chem. Soc. 1995;117:5001–5002.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
