

Microbial imbalance and intestinal pathologies: connections and contributions
- PMID: 25256712
- PMCID: PMC4174524
- DOI: 10.1242/dmm.016428
Microbial imbalance and intestinal pathologies: connections and contributions
Abstract
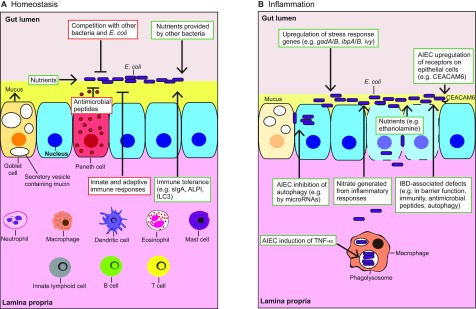
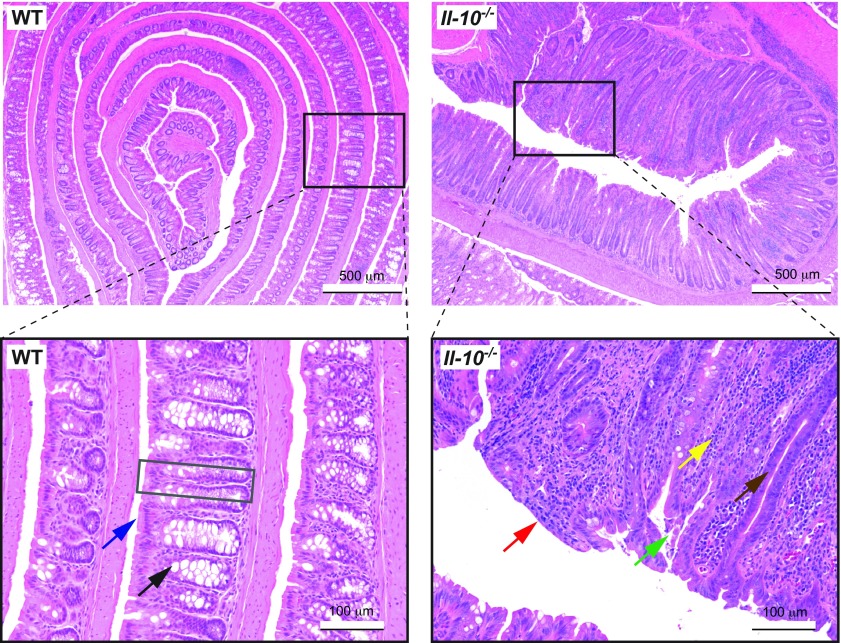
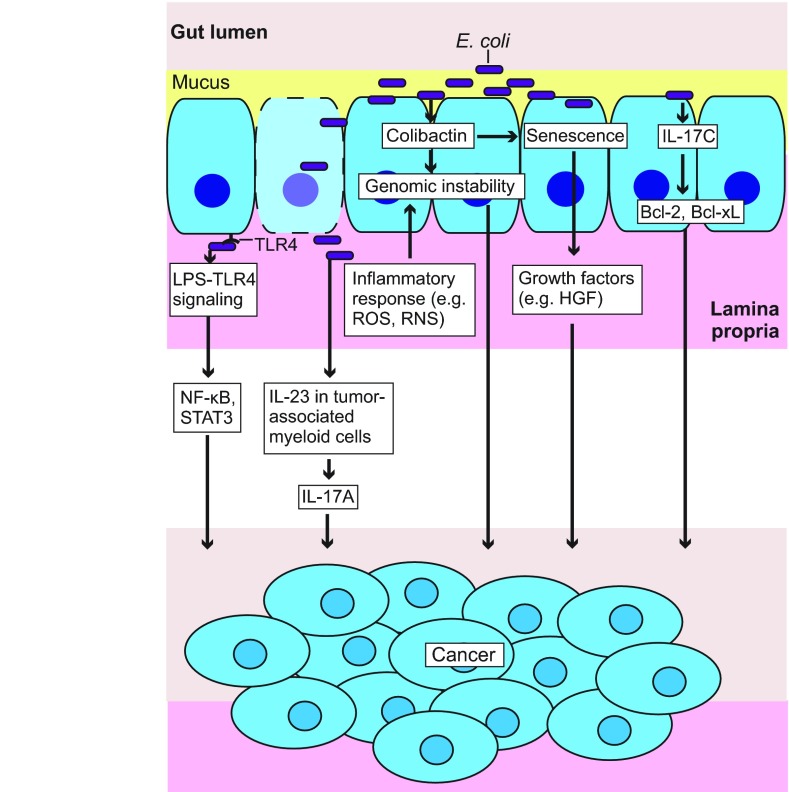
Microbiome analysis has identified a state of microbial imbalance (dysbiosis) in patients with chronic intestinal inflammation and colorectal cancer. The bacterial phylum Proteobacteria is often overrepresented in these individuals, with Escherichia coli being the most prevalent species. It is clear that a complex interplay between the host, bacteria and bacterial genes is implicated in the development of these intestinal diseases. Understanding the basic elements of these interactions could have important implications for disease detection and management. Recent studies have revealed that E. coli utilizes a complex arsenal of virulence factors to colonize and persist in the intestine. Some of these virulence factors, such as the genotoxin colibactin, were found to promote colorectal cancer in experimental models. In this Review, we summarize key features of the dysbiotic states associated with chronic intestinal inflammation and colorectal cancer, and discuss how the dysregulated interplay between host and bacteria could favor the emergence of E. coli with pathological traits implicated in these pathologies.
Keywords: Adherent-invasive E. coli; CRC; Colibactin; Dysbiosis; IBD.
© 2014. Published by The Company of Biologists Ltd.
Figures
References
-
- Alpert C., Scheel J., Engst W., Loh G., Blaut M. (2009). Adaptation of protein expression by Escherichia coli in the gastrointestinal tract of gnotobiotic mice. Environ. Microbiol. 11, 751–761 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
