

Genomic repeat abundances contain phylogenetic signal
- PMID: 25261464
- PMCID: PMC4265144
- DOI: 10.1093/sysbio/syu080
Genomic repeat abundances contain phylogenetic signal
Abstract
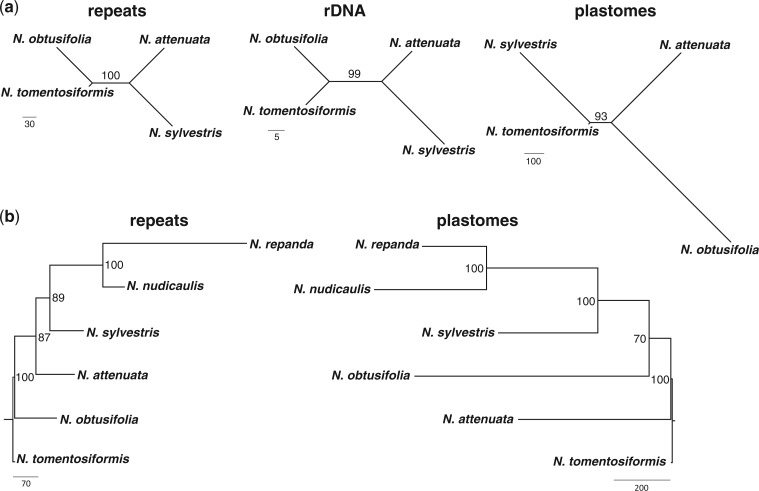
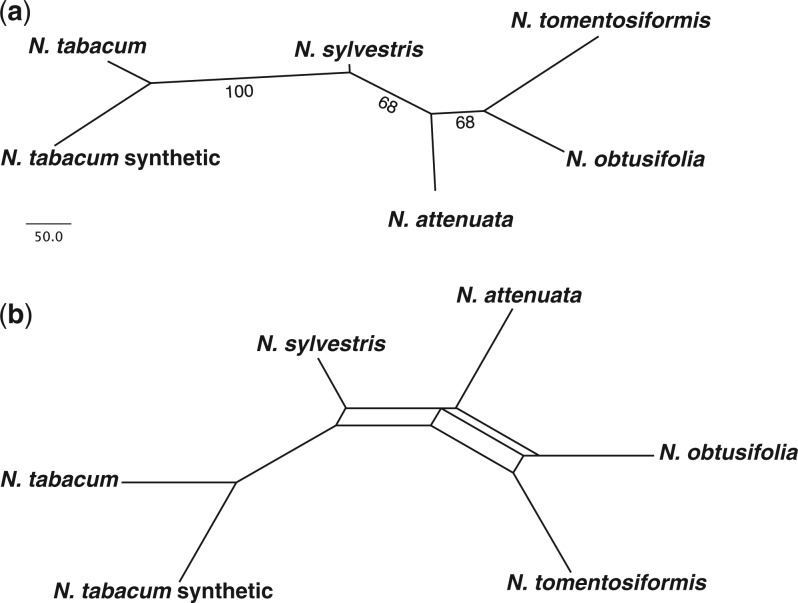
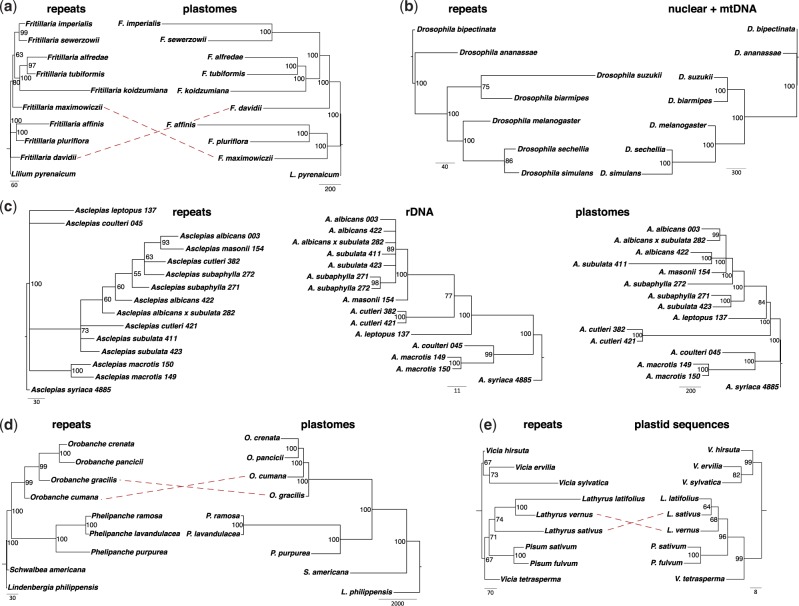
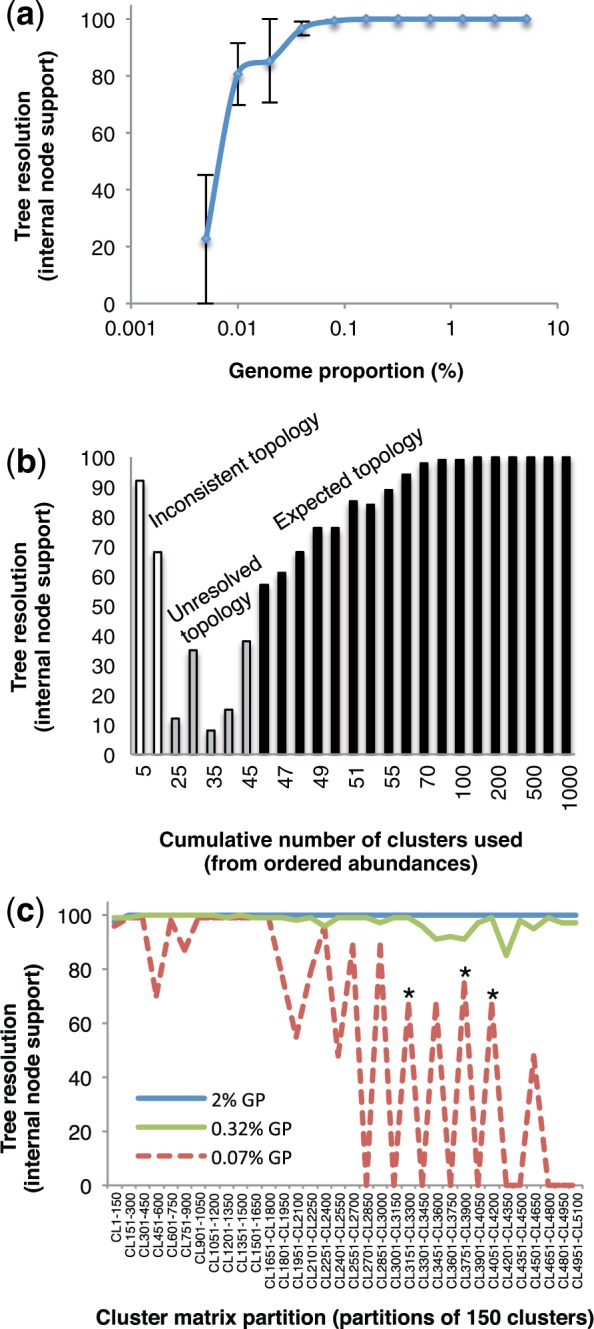
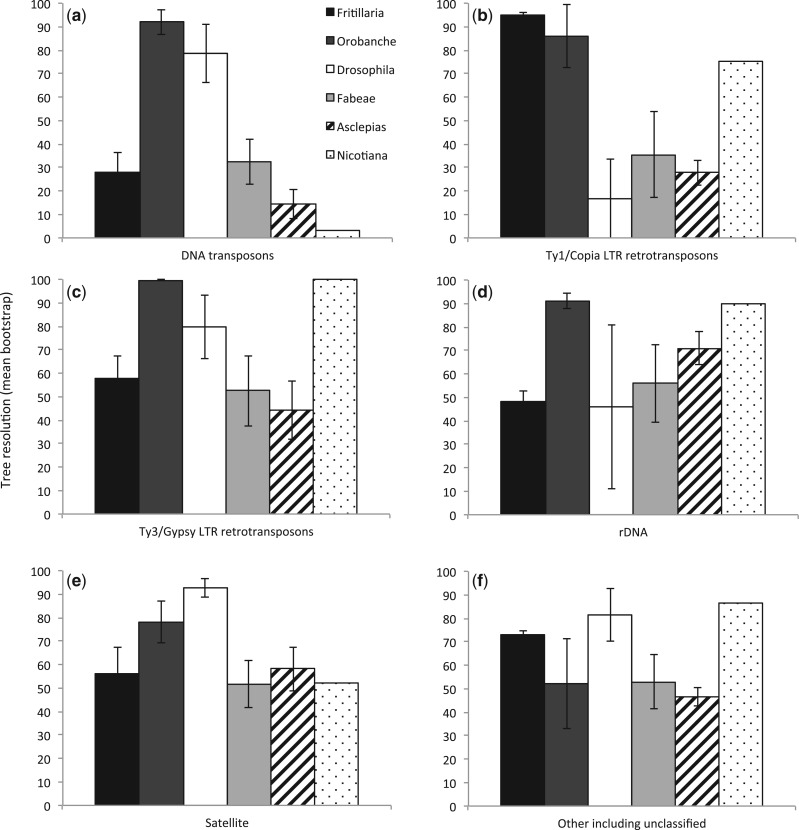
A large proportion of genomic information, particularly repetitive elements, is usually ignored when researchers are using next-generation sequencing. Here we demonstrate the usefulness of this repetitive fraction in phylogenetic analyses, utilizing comparative graph-based clustering of next-generation sequence reads, which results in abundance estimates of different classes of genomic repeats. Phylogenetic trees are then inferred based on the genome-wide abundance of different repeat types treated as continuously varying characters; such repeats are scattered across chromosomes and in angiosperms can constitute a majority of nuclear genomic DNA. In six diverse examples, five angiosperms and one insect, this method provides generally well-supported relationships at interspecific and intergeneric levels that agree with results from more standard phylogenetic analyses of commonly used markers. We propose that this methodology may prove especially useful in groups where there is little genetic differentiation in standard phylogenetic markers. At the same time as providing data for phylogenetic inference, this method additionally yields a wealth of data for comparative studies of genome evolution.
Keywords: Repetitive DNA; continuous characters; genomics; molecular systematics; next-generation sequencing; phylogenetics.
© The Author(s) 2014. Published by Oxford University Press, on behalf of the Society of Systematic Biologists.
Figures
References
-
- Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 2009;161:105–121.
-
- Barrett C.F., Davis J.I., Leebens-Mack J., Conran J.G., Stevenson D.W. Plastid genomes and deep relationships among the commelinid monocot angiosperms. Cladistics. 2013;29:65–87. - PubMed
-
- Bock D.G., Kane N.C., Ebert D.P., Rieseberg L.H. Genome skimming reveals the origin of the Jerusalem Artichoke tuber crop species: neither from Jerusalem nor an artichoke. New Phyt. 2014;201:1021–1030. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
