

Mitochondrial dysfunction induced by a post-translationally modified amyloid linked to a familial mutation in an alternative model of neurodegeneration
- PMID: 25261792
- PMCID: PMC4454292
- DOI: 10.1016/j.bbadis.2014.09.010
Mitochondrial dysfunction induced by a post-translationally modified amyloid linked to a familial mutation in an alternative model of neurodegeneration
Abstract
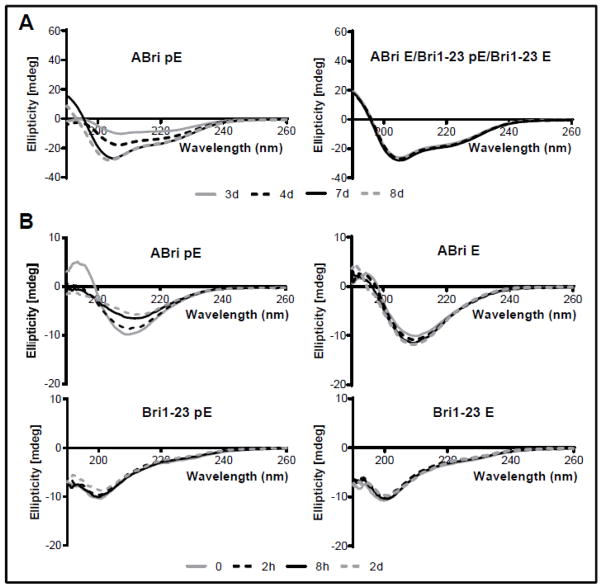
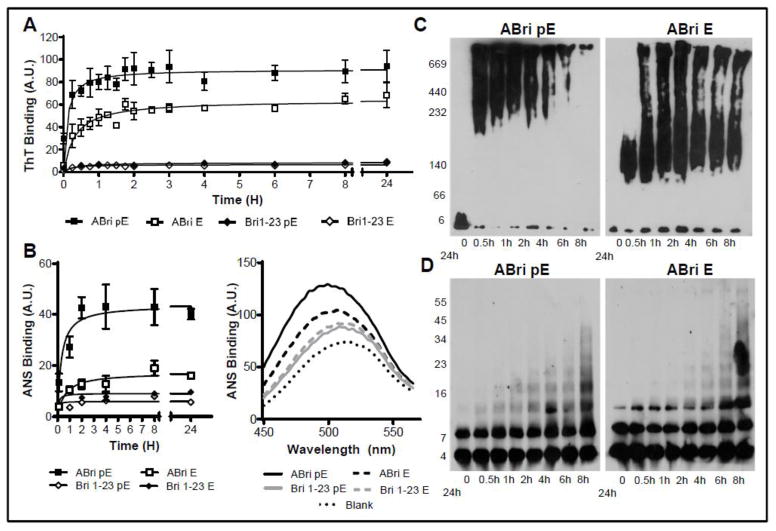
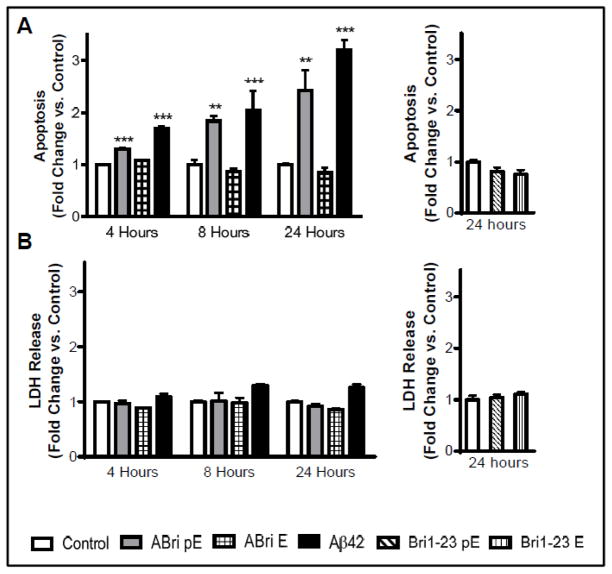
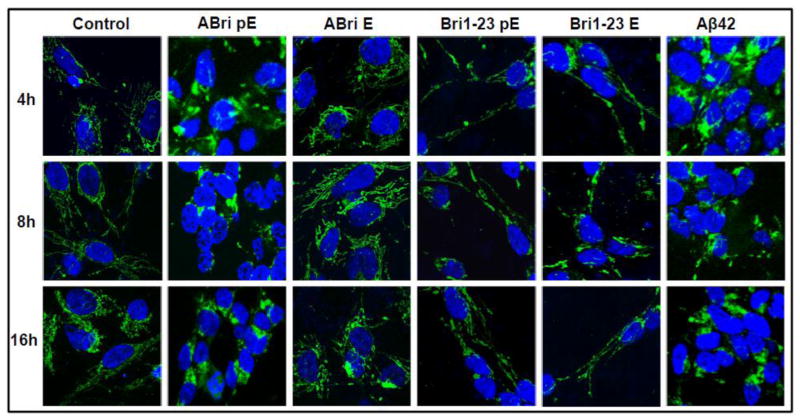
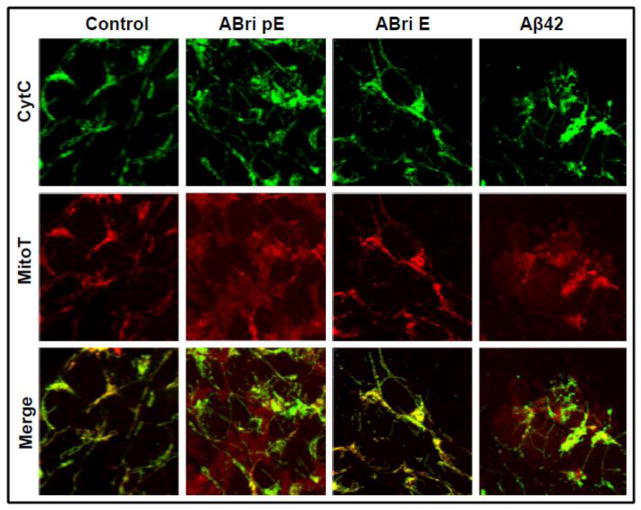
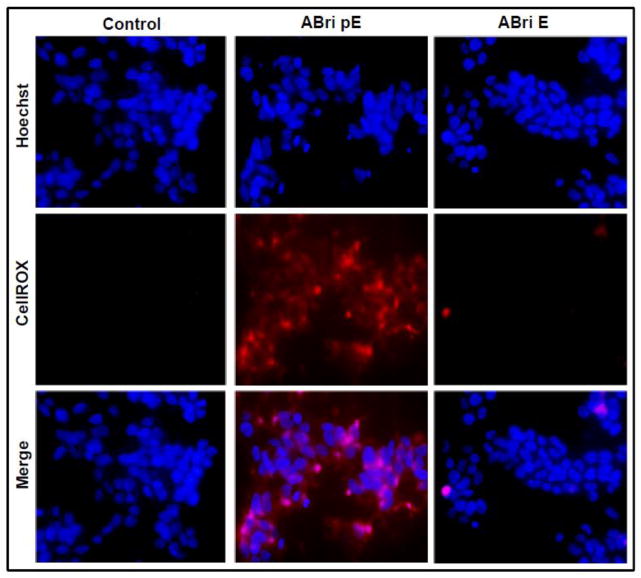
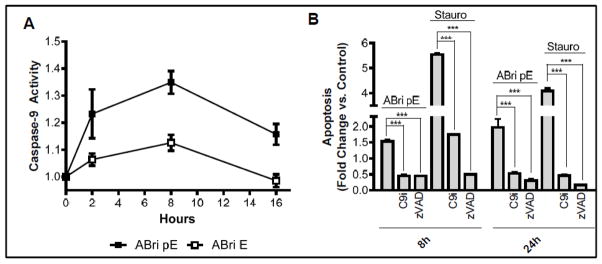
Familial British dementia (FBD) is an early-onset non-amyloid-β (Aβ) cerebral amyloidosis that presents with severe cognitive decline and strikingly similar neuropathological features to those present in Alzheimer's disease (AD). FBD is associated with a T to A single nucleotide transition in the stop codon of a gene encoding BRI2, leading to the production of an elongated precursor protein. Furin-like proteolytic processing at its C-terminus releases a longer-than-normal 34 amino acid peptide, ABri, exhibiting amyloidogenic properties not seen in its 23 amino acid physiologic counterpart Bri1-23. Deposited ABri exhibits abundant post-translational pyroglutamate (pE) formation at the N-terminus, a feature seen in truncated forms of Aβ found in AD deposits, and co-exists with neurofibrillary tangles almost identical to those found in AD. We tested the impact of the FBD mutation alone and in conjunction with the pE post-translational modification on the structural properties and associated neurotoxicity of the ABri peptide. The presence of pE conferred to the ABri molecule enhanced hydrophobicity and accelerated aggregation/fibrillization properties. ABri pE was capable of triggering oxidative stress, loss of mitochondrial membrane potential and activation of caspase-mediated apoptotic mechanisms in neuronal cells, whereas homologous peptides lacking the elongated C-terminus and/or the N-terminal pE were unable to induce similar detrimental cellular pathways. The data indicate that the presence of N-terminal pE is not in itself sufficient to induce pathogenic changes in the physiologic Bri1-23 peptides but that its combination with the ABri mutation is critical for the molecular pathogenesis of FBD.
Keywords: Apoptosis; Cerebral amyloidosis; Cytochrome c; Familial British dementia; Oligomeric amyloid assemblies; Oxidative stress.
Copyright © 2014 Elsevier B.V. All rights reserved.
Figures
Similar articles
-
Chemical traits of cerebral amyloid angiopathy in familial British-, Danish-, and non-Alzheimer's dementias.J Neurochem. 2022 Nov;163(3):233-246. doi: 10.1111/jnc.15694. Epub 2022 Sep 25. J Neurochem. 2022. PMID: 36102248 Free PMC article.
-
The Familial British Dementia Mutation Promotes Formation of Neurotoxic Cystine Cross-linked Amyloid Bri (ABri) Oligomers.J Biol Chem. 2015 Jul 3;290(27):16502-16. doi: 10.1074/jbc.M115.652263. Epub 2015 May 8. J Biol Chem. 2015. PMID: 25957407 Free PMC article.
-
Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial Danish dementia ADan amyloid.Neurobiol Dis. 2016 Jan;85:130-143. doi: 10.1016/j.nbd.2015.10.003. Epub 2015 Oct 13. Neurobiol Dis. 2016. PMID: 26459115 Free PMC article.
-
Chromosome 13 dementias.Cell Mol Life Sci. 2005 Aug;62(16):1814-25. doi: 10.1007/s00018-005-5092-5. Cell Mol Life Sci. 2005. PMID: 15968464 Free PMC article. Review.
-
Chromosome 13 dementia syndromes as models of neurodegeneration.Amyloid. 2001 Dec;8(4):277-84. doi: 10.3109/13506120108993826. Amyloid. 2001. PMID: 11791622 Review.
Cited by
-
RNA Expression Profile and Potential Biomarkers in Patients With Spinocerebellar Ataxia Type 3 From Mainland China.Front Genet. 2019 Jun 13;10:566. doi: 10.3389/fgene.2019.00566. eCollection 2019. Front Genet. 2019. PMID: 31249598 Free PMC article.
-
Potentiation of neurotoxicity in double-mutant mice with Pink1 ablation and A53T-SNCA overexpression.Hum Mol Genet. 2015 Feb 15;24(4):1061-76. doi: 10.1093/hmg/ddu520. Epub 2014 Oct 8. Hum Mol Genet. 2015. PMID: 25296918 Free PMC article.
-
Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer's Disease Pathogenesis.Front Aging Neurosci. 2019 Nov 20;11:311. doi: 10.3389/fnagi.2019.00311. eCollection 2019. Front Aging Neurosci. 2019. PMID: 31824296 Free PMC article. Review.
-
N-terminally truncated Aβ4-x proteoforms and their relevance for Alzheimer's pathophysiology.Transl Neurodegener. 2022 Jun 1;11(1):30. doi: 10.1186/s40035-022-00303-3. Transl Neurodegener. 2022. PMID: 35641972 Free PMC article.
-
Aβ truncated species: Implications for brain clearance mechanisms and amyloid plaque deposition.Biochim Biophys Acta Mol Basis Dis. 2018 Jan;1864(1):208-225. doi: 10.1016/j.bbadis.2017.07.005. Epub 2017 Jul 13. Biochim Biophys Acta Mol Basis Dis. 2018. PMID: 28711595 Free PMC article.
References
-
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiological Reviews. 2001;81:741–766. - PubMed
-
- Ghiso J, Frangione B. Cerebral amyloidosis, amyloid angiopathy, and their relationship to stroke and dementia. J Alzheimers Dis. 2001;3:65–73. - PubMed
-
- Querfurth H, LaFerla F. Alzheimer’s disease. The New England journal of medicine. 2010;362:329–344. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
