

Genome-wide analysis of noncoding regulatory mutations in cancer
- PMID: 25261935
- PMCID: PMC4217527
- DOI: 10.1038/ng.3101
Genome-wide analysis of noncoding regulatory mutations in cancer
Abstract
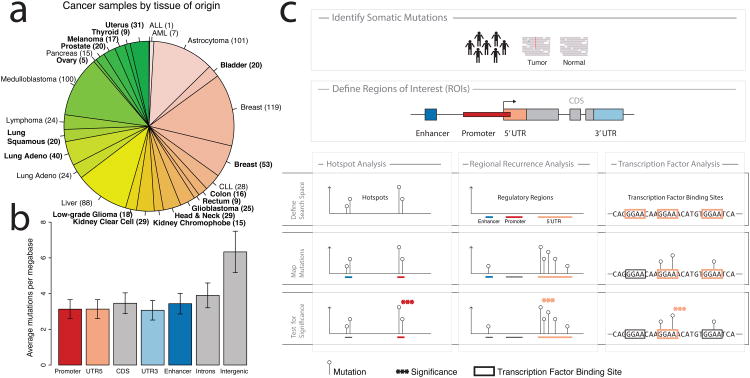
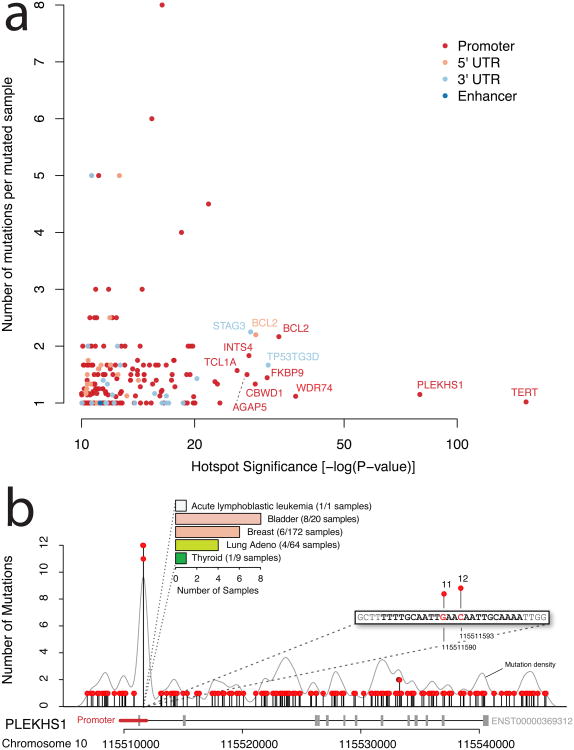
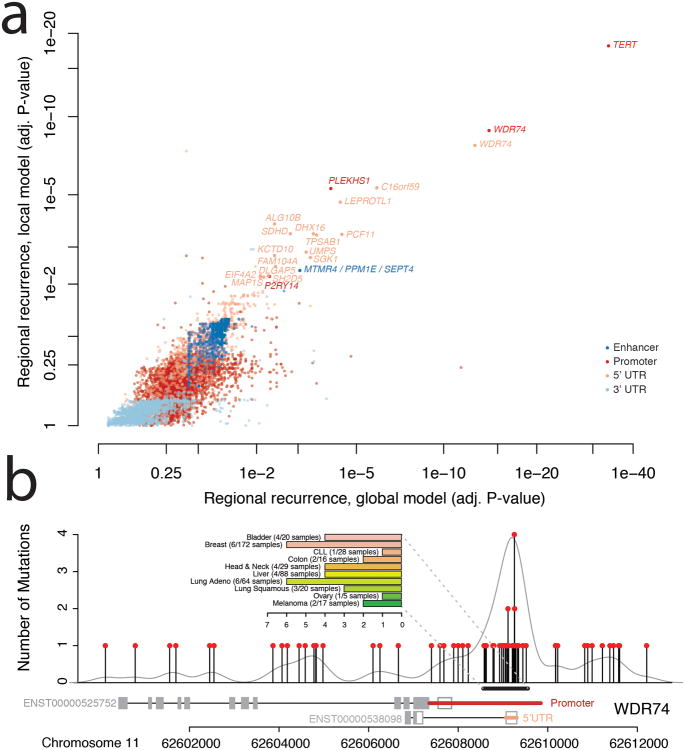
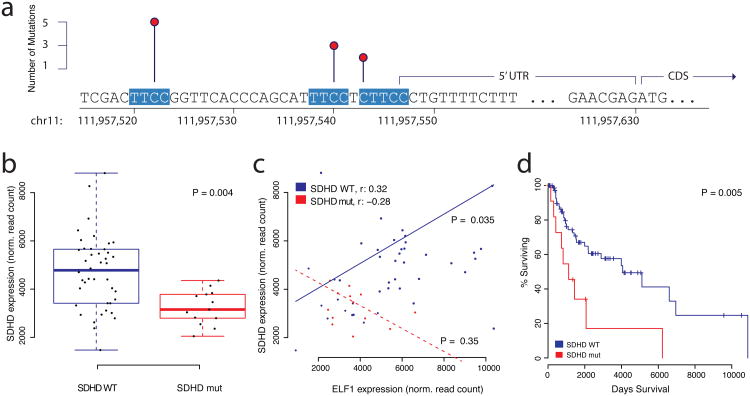
Cancer primarily develops because of somatic alterations in the genome. Advances in sequencing have enabled large-scale sequencing studies across many tumor types, emphasizing the discovery of alterations in protein-coding genes. However, the protein-coding exome comprises less than 2% of the human genome. Here we analyze the complete genome sequences of 863 human tumors from The Cancer Genome Atlas and other sources to systematically identify noncoding regions that are recurrently mutated in cancer. We use new frequency- and sequence-based approaches to comprehensively scan the genome for noncoding mutations with potential regulatory impact. These methods identify recurrent mutations in regulatory elements upstream of PLEKHS1, WDR74 and SDHD, as well as previously identified mutations in the TERT promoter. SDHD promoter mutations are frequent in melanoma and are associated with reduced gene expression and poor prognosis. The non-protein-coding cancer genome remains widely unexplored, and our findings represent a step toward targeting the entire genome for clinical purposes.
Conflict of interest statement
Figures
Comment in
-
Second call for pan-cancer analysis.Nat Genet. 2014 Dec;46(12):1251. doi: 10.1038/ng.3155. Nat Genet. 2014. PMID: 25418742 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
