

Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor α-positive breast cancer
- PMID: 25263449
- PMCID: PMC4377305
- DOI: 10.1038/onc.2014.292
Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor α-positive breast cancer
Abstract
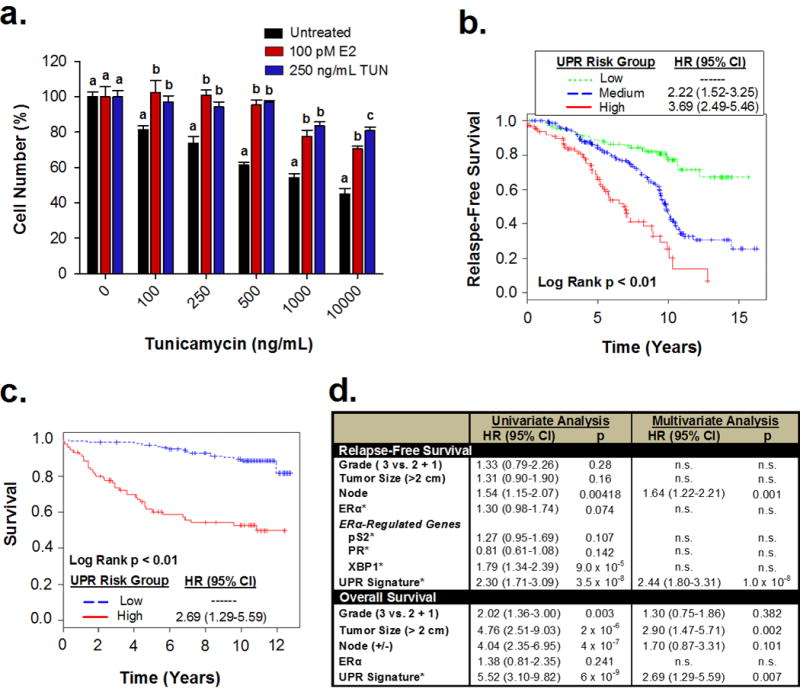
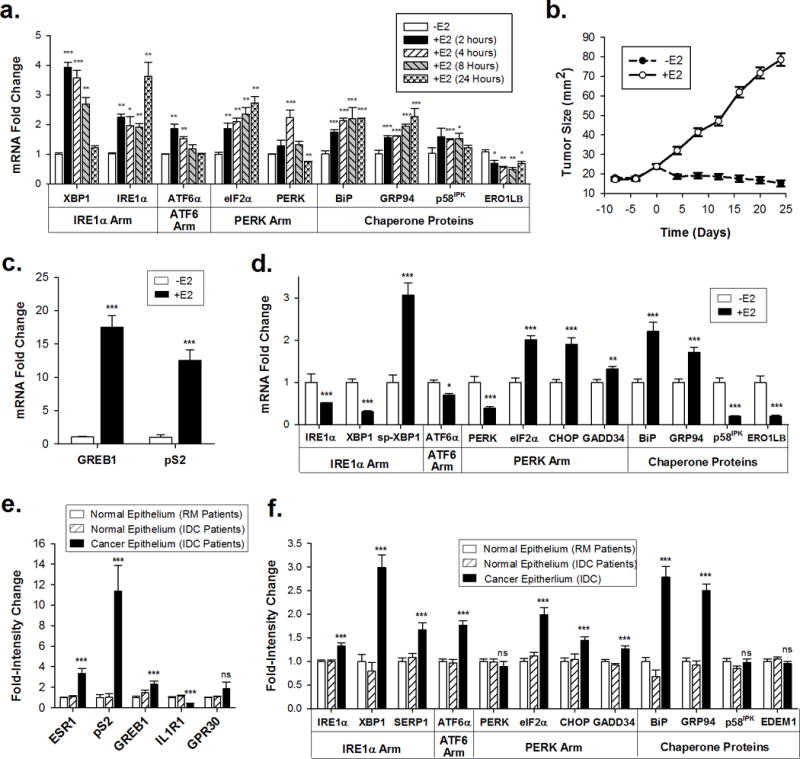
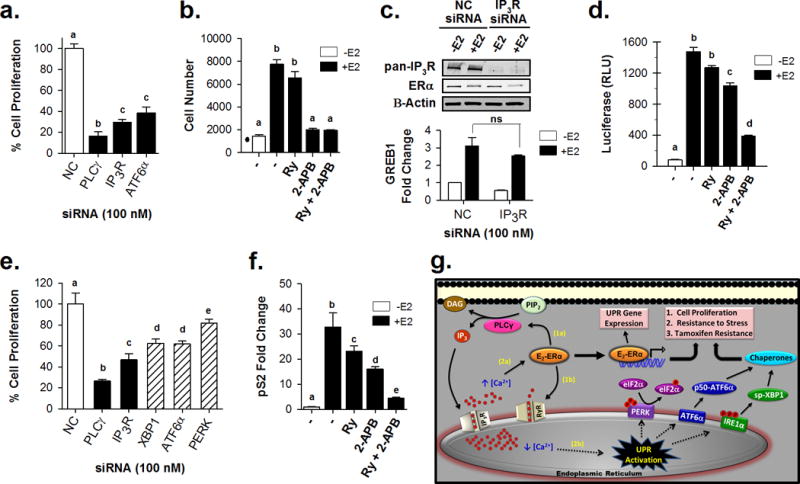
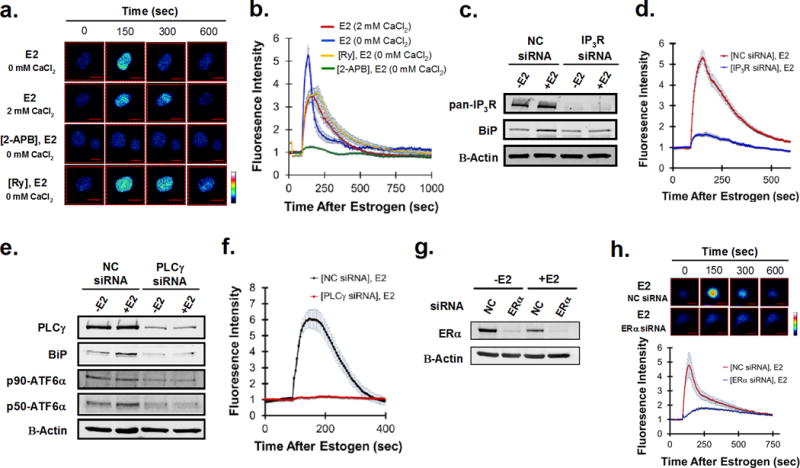
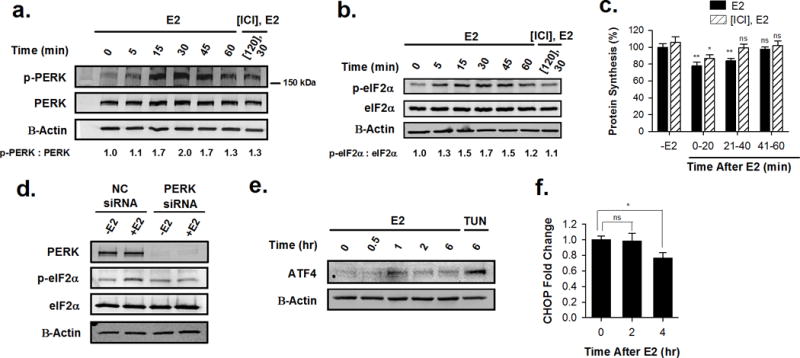
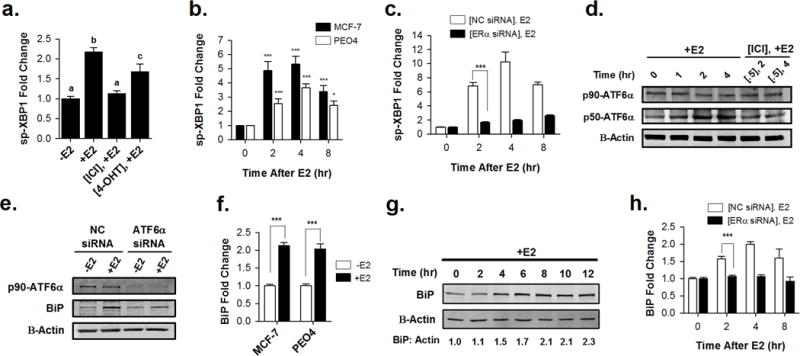
In response to cell stress, cancer cells often activate the endoplasmic reticulum (EnR) stress sensor, the unfolded protein response (UPR). Little was known about the potential role in cancer of a different mode of UPR activation, anticipatory activation of the UPR prior to accumulation of unfolded protein or cell stress. We show that estrogen, acting via estrogen receptor α (ERα), induces rapid anticipatory activation of the UPR, resulting in increased production of the antiapoptotic chaperone BiP/GRP78, preparing cancer cells for the increased protein production required for subsequent estrogen-ERα-induced cell proliferation. In ERα-containing cancer cells, the estrogen, 17β-estradiol (E2) activates the UPR through a phospholipase C γ (PLCγ)-mediated opening of EnR IP3R calcium channels, enabling passage of calcium from the lumen of the EnR into the cytosol. siRNA knockdown of ERα blocked the estrogen-mediated increase in cytosol calcium and UPR activation. Knockdown or inhibition of PLCγ, or of IP3R, strongly inhibited the estrogen-mediated increases in cytosol calcium, UPR activation and cell proliferation. E2-ERα activates all three arms of the UPR in breast and ovarian cancer cells in culture and in a mouse xenograft. Knockdown of ATF6α, which regulates UPR chaperones, blocked estrogen induction of BiP and strongly inhibited E2-ERα-stimulated cell proliferation. Mild and transient UPR activation by estrogen promotes an adaptive UPR response that protects cells against subsequent UPR-mediated apoptosis. Analysis of data from ERα(+) breast cancers demonstrates elevated expression of a UPR gene signature that is a powerful new prognostic marker tightly correlated with subsequent resistance to tamoxifen therapy, reduced time to recurrence and poor survival. Thus, as an early component of the E2-ERα proliferation program, the mitogen estrogen, drives rapid anticipatory activation of the UPR. Anticipatory activation of the UPR is a new role for estrogens in cancer cell proliferation and resistance to therapy.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–643. - PubMed
-
- Yue W, Yager JD, Wang JP, Jupe ER, Santen RJ. Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids. 2013;78:161–170. - PubMed
-
- (EBCTCG). EBCTCG. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. - PubMed
-
- Jensen EV, Jordan VC. The estrogen receptor: a model for molecular medicine. Clin Cancer Res. 2003;9:1980–1989. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
