

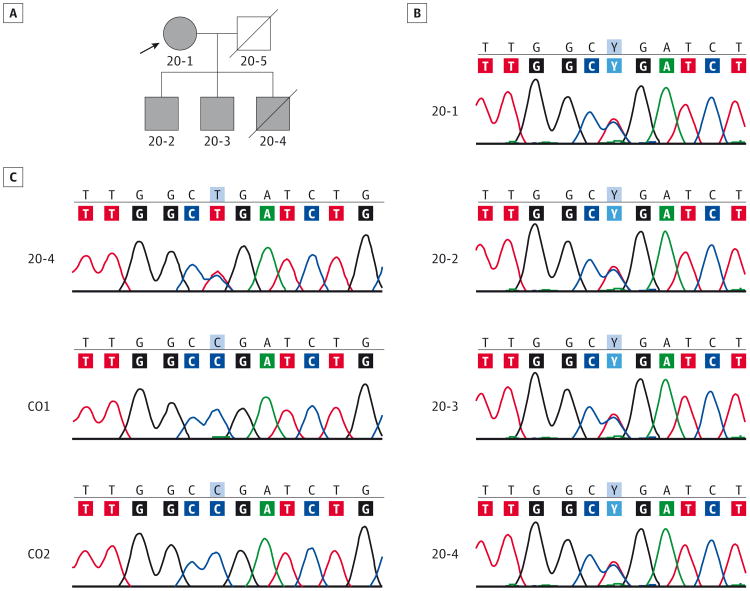
Expanding the phenotype associated with the NEFL mutation: neuromuscular disease in a family with overlapping myopathic and neurogenic findings
- PMID: 25264603
- PMCID: PMC4227917
- DOI: 10.1001/jamaneurol.2014.1432
Expanding the phenotype associated with the NEFL mutation: neuromuscular disease in a family with overlapping myopathic and neurogenic findings
Abstract
Importance: Newer sequencing technologies in combination with traditional gene mapping techniques, such as linkage analysis, can help identify the genetic basis of disease for patients with rare disorders of uncertain etiology. This approach may expand the phenotypic spectrum of disease associated with those genetic mutations.
Objective: To elucidate the molecular cause of a neuromuscular disease among a family in which 4 members, a mother and her 3 sons, were affected.
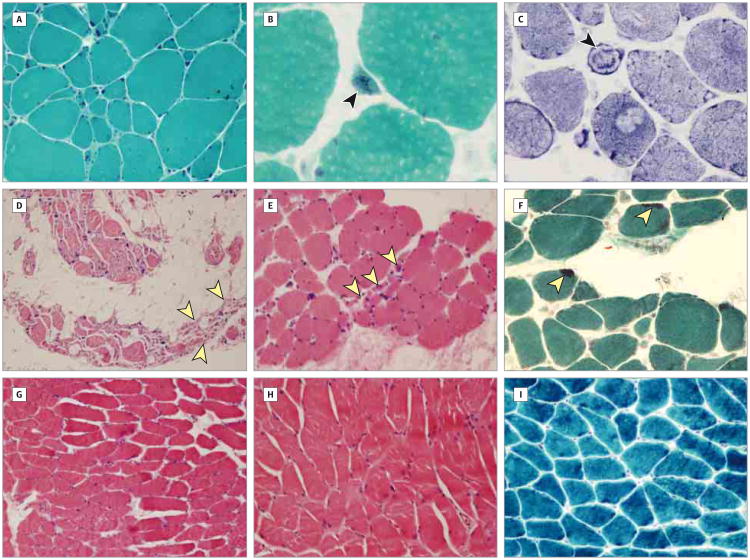
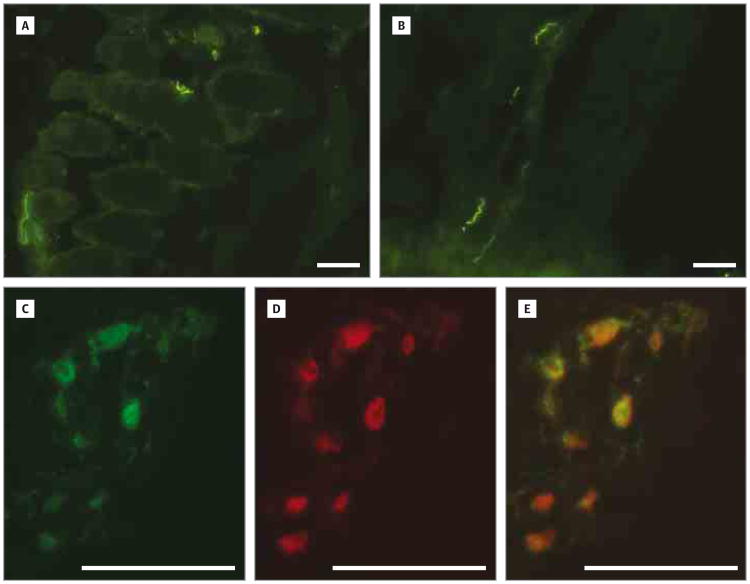
Design, setting, and participants: Two of 4 affected members manifested nemaline myopathy, a common subtype of congenital myopathy, while the other 2 had a nonspecific myopathy. Single-nucleotide polymorphism-based linkage analysis was performed on DNA samples from the 4 affected family members, and whole-genome sequencing was performed in the proband. Real-time quantitative reverse transcription-polymerase chain reaction, immunofluorescence, and Western blot analysis were performed on muscle biopsy specimens.
Main outcomes and measures: Whole-genome sequencing and linkage analysis identified a variant in a gene that explains the phenotype.
Results: We identified a novel neurofilament light polypeptide (NEFL) nonsense mutation in all affected members. NEFL mutations have been previously linked to Charcot-Marie-Tooth disease in humans. This led us to reevaluate the diagnosis, and we recognized that several of the findings, especially those related to the muscle biopsy specimens and electromyography, were consistent with a neurogenic disease.
Conclusions and relevance: NEFL mutations are known to cause Charcot-Marie-Tooth disease in humans and motor neuron disease in mice. We report the identification of an NEFL mutation in a family clinically manifesting congenital myopathy. We also describe potential overlap between myopathic and neurogenic findings in this family. These findings expand the phenotypic spectrum of diseases associated with NEFL mutations. This study is an example of the power of genomic approaches to identify potentially pathogenic mutations in unsuspected genes responsible for heterogeneous neuromuscular diseases.
Conflict of interest statement
Figures
References
-
- Komlósi K, Hadzsiev K, Garbes L, et al. Exome sequencing identifies Laing distal myopathy MYH7 mutation in a Roma family previously diagnosed with distal neuronopathy. Neuromuscul Disord. 2014;24(2):156–161. - PubMed
-
- Igari R, Wada M, Sato H, Hayashi KY, Nishino I, Kato T. A case of inclusion body myopathy with Paget's disease of bone and frontotemporal dementia (IBMPFD) showing clinical features of motor neuron disease. Rinsho Shinkeigaku. 2013;53(6):458–464. in Japanese. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
