

Polycystic liver diseases: advanced insights into the molecular mechanisms
- PMID: 25266109
- PMCID: PMC4526263
- DOI: 10.1038/nrgastro.2014.155
Polycystic liver diseases: advanced insights into the molecular mechanisms
Abstract
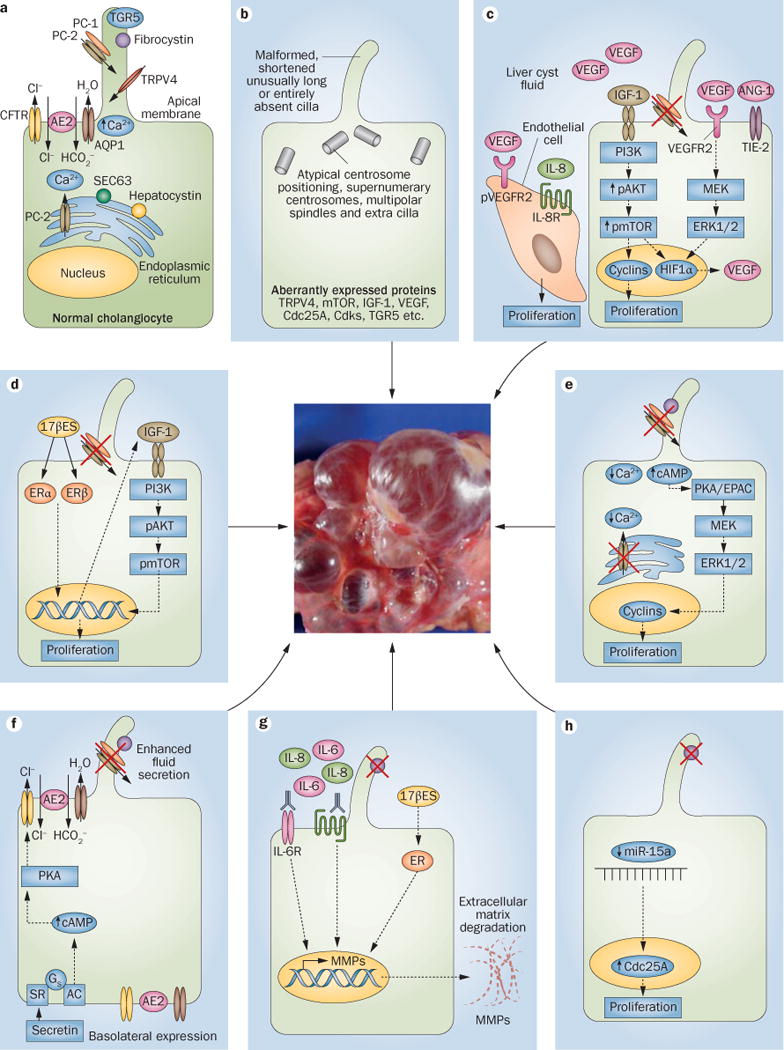
Polycystic liver diseases are genetic disorders characterized by progressive bile duct dilatation and/or cyst development. The large volume of hepatic cysts causes different symptoms and complications such as abdominal distension, local pressure with back pain, hypertension, gastro-oesophageal reflux and dyspnea as well as bleeding, infection and rupture of the cysts. Current therapeutic strategies are based on surgical procedures and pharmacological management, which partially prevent or ameliorate the disease. However, as these treatments only show short-term and/or modest beneficial effects, liver transplantation is the only definitive therapy. Therefore, interest in understanding the molecular mechanisms involved in disease pathogenesis is increasing so that new targets for therapy can be identified. In this Review, the genetic mechanisms underlying polycystic liver diseases and the most relevant molecular pathways of hepatic cystogenesis are discussed. Moreover, the main clinical and preclinical studies are highlighted and future directions in basic as well as clinical research are indicated.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Gevers TJ, Drenth JP. Diagnosis and management of polycystic liver disease. Nat Rev Gastroenterol Hepatol. 2013;10:101–108. - PubMed
-
- Drenth JP, te Morsche RH, Smink R, Bonifacino JS, Jansen JB. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet. 2003;33:345–347. - PubMed
-
- Davila S, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575–577. - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
