

c-Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells
- PMID: 25266721
- PMCID: PMC4231650
- DOI: 10.1074/jbc.M114.574517
c-Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells
Abstract
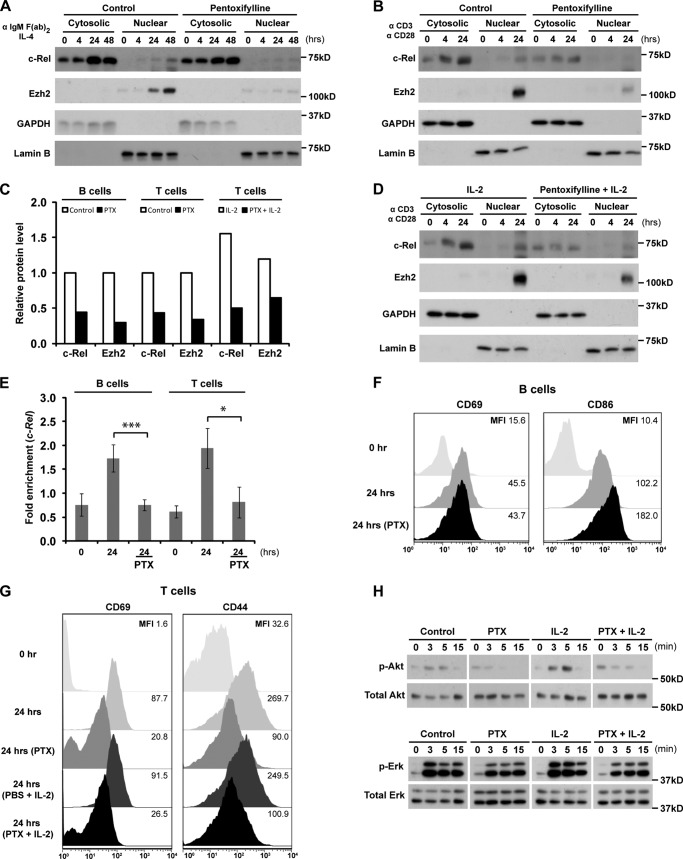
The polycomb group protein Ezh2 is a histone methyltransferase that modifies chromatin structure to alter gene expression during embryonic development, lymphocyte activation, and tumorigenesis. The mechanism by which Ezh2 expression is regulated is not well defined. In the current study, we report that c-Rel is a critical activator of Ezh2 transcription in lymphoid cells. In activated primary murine B and T cells, plus human leukemia and multiple myeloma cell lines, recruitment of c-Rel to the first intron of the Ezh2 locus promoted Ezh2 mRNA expression. This up-regulation was abolished in activated c-Rel-deficient lymphocytes and by c-Rel knockdown in Jurkat T cells. Treatment of malignant cells with the c-Rel inhibitor pentoxifylline not only reduced c-Rel nuclear translocation and Ezh2 expression, but also enhanced their sensitivity to the Ezh2-specific drug, GSK126 through increased growth inhibition and cell death. In summary, our demonstration that c-Rel regulates Ezh2 expression in lymphocytes and malignant lymphoid cells reveals a novel transcriptional network in transformed lymphoid cells expressing high levels of Ezh2 that provides a molecular justification for combinatorial drug therapy.
Keywords: Gene Regulation; Leukemia; Lymphocyte; Lymphoma; Polycomb; Transcription Factor.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures







Similar articles
-
EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations.Nature. 2012 Dec 6;492(7427):108-12. doi: 10.1038/nature11606. Epub 2012 Oct 10. Nature. 2012. PMID: 23051747
-
Targeting activating mutations of EZH2 leads to potent cell growth inhibition in human melanoma by derepression of tumor suppressor genes.Oncotarget. 2015 Sep 29;6(29):27023-36. doi: 10.18632/oncotarget.4809. Oncotarget. 2015. PMID: 26304929 Free PMC article.
-
[Effects and molecular mechanism of histone methyltransferase enhancer of zeste homolog 2 on regulating sepsis-induced T cell dysfunction].Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2022 Apr;34(4):357-361. doi: 10.3760/cma.j.cn121430-20210906-01323. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2022. PMID: 35692198 Chinese.
-
EZH2 in normal hematopoiesis and hematological malignancies.Oncotarget. 2016 Jan 19;7(3):2284-96. doi: 10.18632/oncotarget.6198. Oncotarget. 2016. PMID: 26497210 Free PMC article. Review.
-
EZH2 abnormalities in lymphoid malignancies: underlying mechanisms and therapeutic implications.J Hematol Oncol. 2019 Nov 21;12(1):118. doi: 10.1186/s13045-019-0814-6. J Hematol Oncol. 2019. PMID: 31752930 Free PMC article. Review.
Cited by
-
Epigenetic reprogramming of T cells: unlocking new avenues for cancer immunotherapy.Cancer Metastasis Rev. 2024 Mar;43(1):175-195. doi: 10.1007/s10555-024-10167-w. Epub 2024 Jan 18. Cancer Metastasis Rev. 2024. PMID: 38233727 Review.
-
The biological significance of histone modifiers in multiple myeloma: clinical applications.Blood Cancer J. 2018 Aug 22;8(9):83. doi: 10.1038/s41408-018-0119-y. Blood Cancer J. 2018. PMID: 30190472 Free PMC article. Review.
-
c-Rel orchestrates energy-dependent epithelial and macrophage reprogramming in fibrosis.Nat Metab. 2020 Nov;2(11):1350-1367. doi: 10.1038/s42255-020-00306-2. Epub 2020 Nov 9. Nat Metab. 2020. PMID: 33168981 Free PMC article.
-
Genetic Predictors of Mortality in Patients with Multiple Myeloma.Appl Clin Genet. 2021 Apr 29;14:241-254. doi: 10.2147/TACG.S262866. eCollection 2021. Appl Clin Genet. 2021. PMID: 33953598 Free PMC article. Review.
-
Chemotherapy with a molecular rational basis, pentoxifylline as a promising antitumor drug.Ann Med Surg (Lond). 2025 Feb 28;87(3):1506-1528. doi: 10.1097/MS9.0000000000003043. eCollection 2025 Mar. Ann Med Surg (Lond). 2025. PMID: 40213176 Free PMC article. Review.
References
-
- Lewis E. B. (1978) A gene complex controlling segmentation in Drosophila. Nature 276, 565–570 - PubMed
-
- Czermin B., Melfi R., McCabe D., Seitz V., Imhof A., Pirrotta V. (2002) Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal polycomb sites. Cell 111, 185–196 - PubMed
-
- Francis N. J., Saurin A. J., Shao Z., Kingston R. E. (2001) Reconstitution of a functional core polycomb repressive complex. Mol. Cell 8, 545–556 - PubMed
-
- Müller J., Hart C. M., Francis N. J., Vargas M. L., Sengupta A., Wild B., Miller E. L., O'Connor M. B., Kingston R. E., Simon J. A. (2002) Histone methyltransferase activity of a Drosophila polycomb group repressor complex. Cell 111, 197–208 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
