

The hereditary spastic paraplegia-related enzyme DDHD2 is a principal brain triglyceride lipase
- PMID: 25267624
- PMCID: PMC4205627
- DOI: 10.1073/pnas.1413706111
The hereditary spastic paraplegia-related enzyme DDHD2 is a principal brain triglyceride lipase
Abstract
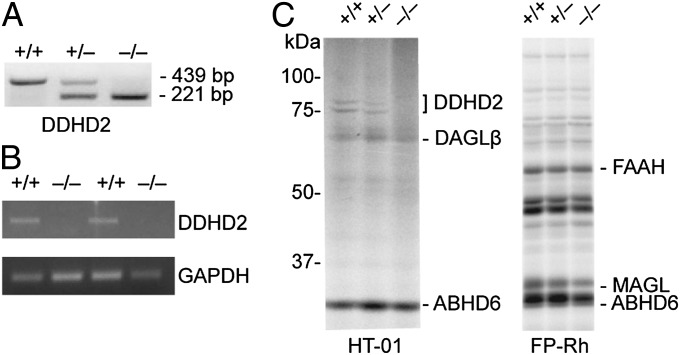
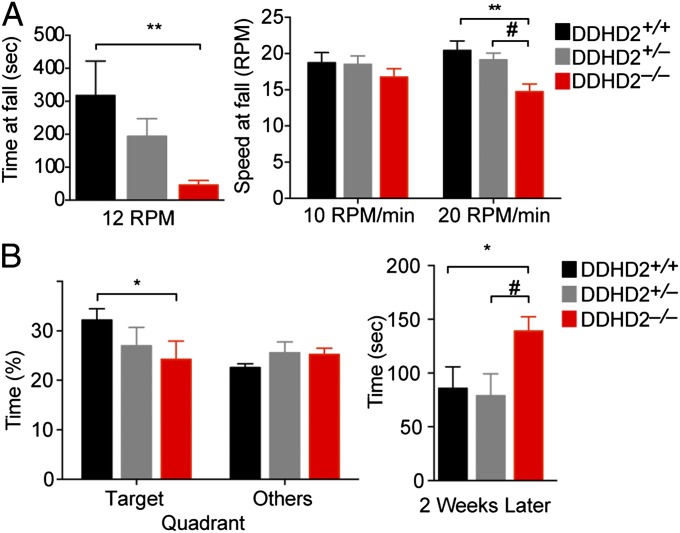
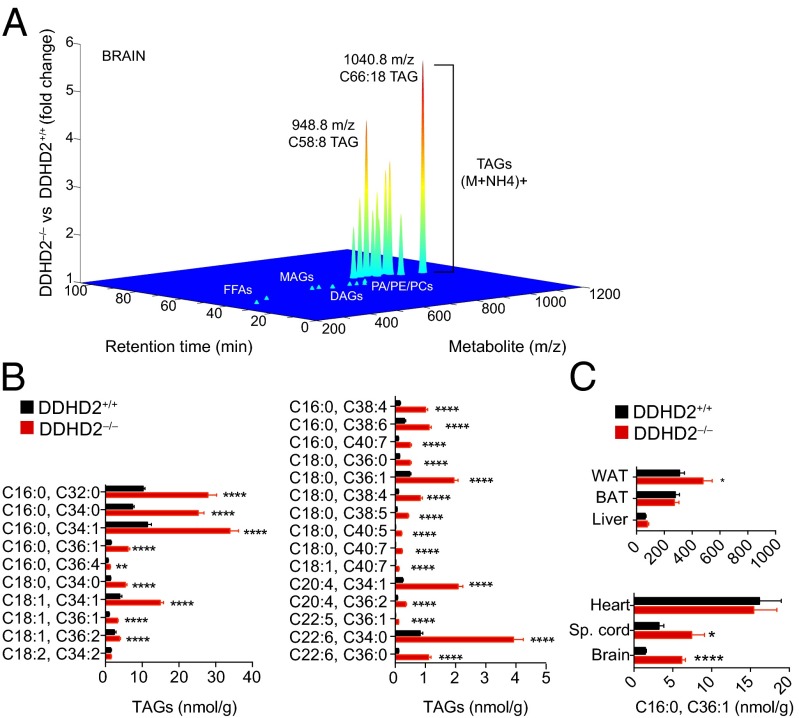
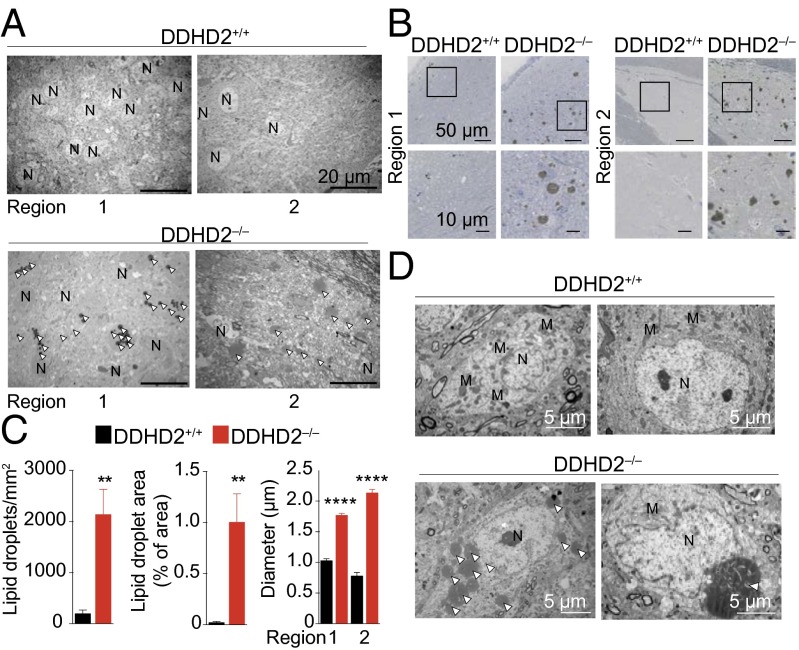
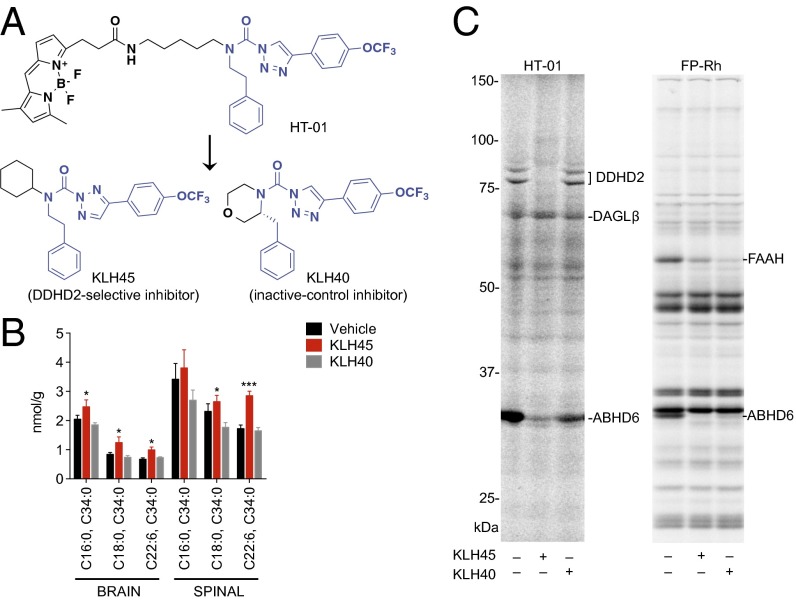
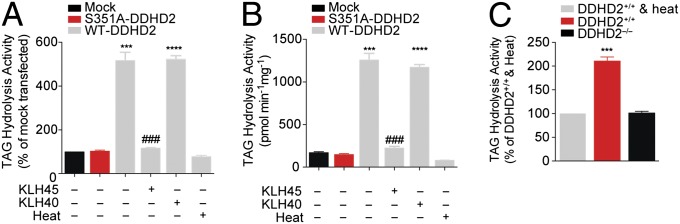
Complex hereditary spastic paraplegia (HSP) is a genetic disorder that causes lower limb spasticity and weakness and intellectual disability. Deleterious mutations in the poorly characterized serine hydrolase DDHD2 are a causative basis for recessive complex HSP. DDHD2 exhibits phospholipase activity in vitro, but its endogenous substrates and biochemical functions remain unknown. Here, we report the development of DDHD2(-/-) mice and a selective, in vivo-active DDHD2 inhibitor and their use in combination with mass spectrometry-based lipidomics to discover that DDHD2 regulates brain triglycerides (triacylglycerols, or TAGs). DDHD2(-/-) mice show age-dependent TAG elevations in the central nervous system, but not in several peripheral tissues. Large lipid droplets accumulated in DDHD2(-/-) brains and were localized primarily to the intracellular compartments of neurons. These metabolic changes were accompanied by impairments in motor and cognitive function. Recombinant DDHD2 displays TAG hydrolase activity, and TAGs accumulated in the brains of wild-type mice treated subchronically with a selective DDHD2 inhibitor. These findings, taken together, indicate that the central nervous system possesses a specialized pathway for metabolizing TAGs, disruption of which leads to massive lipid accumulation in neurons and complex HSP syndrome.
Conflict of interest statement
Conflict of interest statement: The authors declare competing financial interests. B.F.C. is cofounder and advisor for a biotechnology company interested in developing inhibitors for serine hydrolases as therapeutic targets.
Figures
References
-
- Rabbani B, Mahdieh N, Hosomichi K, Nakaoka H, Inoue I. Next-generation sequencing: Impact of exome sequencing in characterizing Mendelian disorders. J Hum Genet. 2012;57(10):621–632. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
