

Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1-KRAS-NF-κB pathway
- PMID: 25267977
- PMCID: PMC4201976
- DOI: 10.4049/jimmunol.1401625
Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1-KRAS-NF-κB pathway
Abstract
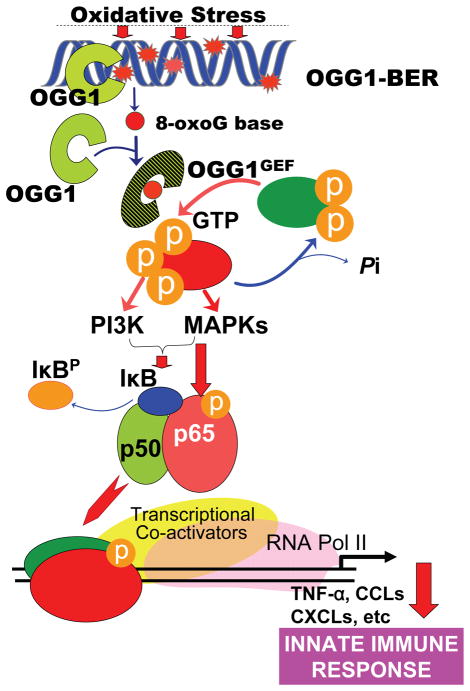
8-Oxoguanine-DNA glycosylase-1 (OGG1) is the primary enzyme for repairing 7,8-dihydro-8-oxoguanine (8-oxoG) via the DNA base excision repair pathway (OGG1-BER). Accumulation of 8-oxoG in the genomic DNA leads to genetic instability and carcinogenesis and is thought to contribute to the worsening of various inflammatory and disease processes. However, the disease mechanism is unknown. In this study, we proposed that the mechanistic link between OGG1-BER and proinflammatory gene expression is OGG1's guanine nucleotide exchange factor activity, acquired after interaction with the 8-oxoG base and consequent activation of the small GTPase RAS. To test this hypothesis, we used BALB/c mice expressing or deficient in OGG1 in their airway epithelium and various molecular biological approaches, including active RAS pulldown, reporter and Comet assays, small interfering RNA-mediated depletion of gene expression, quantitative RT-PCR, and immunoblotting. We report that the OGG1-initiated repair of oxidatively damaged DNA is a prerequisite for GDP → GTP exchange, KRAS-GTP-driven signaling via MAP kinases and PI3 kinases and mitogen-stress-related kinase-1 for NF-κB activation, proinflammatory chemokine/cytokine expression, and inflammatory cell recruitment to the airways. Mice deficient in OGG1-BER showed significantly decreased immune responses, whereas a lack of other Nei-like DNA glycosylases (i.e., NEIL1 and NEIL2) had no significant effect. These data unveil a previously unidentified role of OGG1-driven DNA BER in the generation of endogenous signals for inflammation in the innate signaling pathway.
Copyright © 2014 by The American Association of Immunologists, Inc.
Conflict of interest statement
Figures

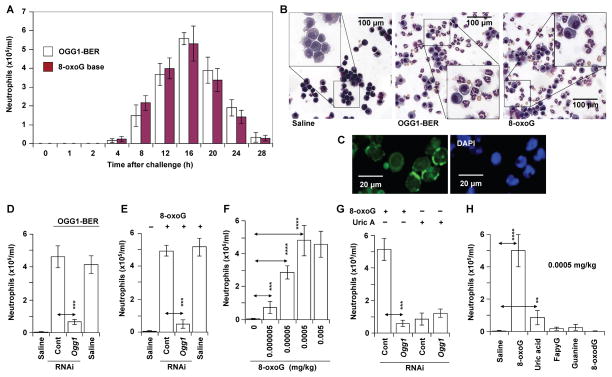
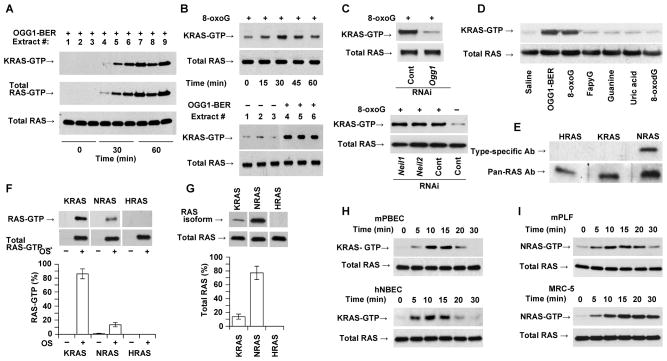
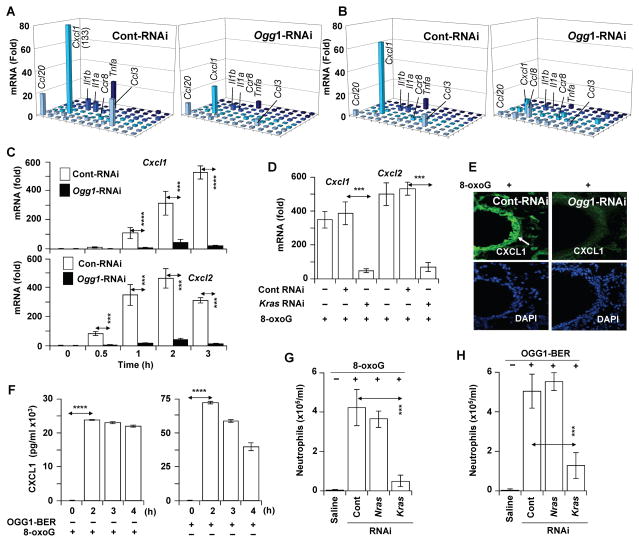
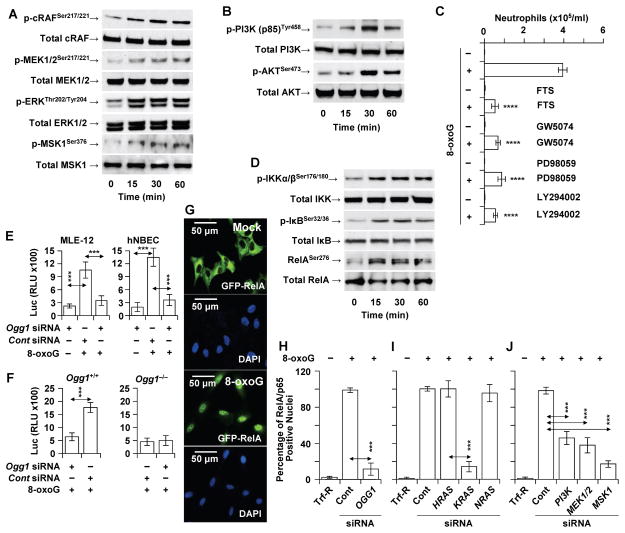
References
-
- D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. - PubMed
-
- Dizdaroglu M. Oxidative damage to DNA in mammalian chromatin. Mutat Res. 1992;275:331–342. - PubMed
-
- Steenken S, Jovanovic SV. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J Am Chem Soc. 1997;119:617–618.
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
