

A universal protocol to generate consensus level genome sequences for foot-and-mouth disease virus and other positive-sense polyadenylated RNA viruses using the Illumina MiSeq
- PMID: 25269623
- PMCID: PMC4247156
- DOI: 10.1186/1471-2164-15-828
A universal protocol to generate consensus level genome sequences for foot-and-mouth disease virus and other positive-sense polyadenylated RNA viruses using the Illumina MiSeq
Abstract
Background: Next-Generation Sequencing (NGS) is revolutionizing molecular epidemiology by providing new approaches to undertake whole genome sequencing (WGS) in diagnostic settings for a variety of human and veterinary pathogens. Previous sequencing protocols have been subject to biases such as those encountered during PCR amplification and cell culture, or are restricted by the need for large quantities of starting material. We describe here a simple and robust methodology for the generation of whole genome sequences on the Illumina MiSeq. This protocol is specific for foot-and-mouth disease virus (FMDV) or other polyadenylated RNA viruses and circumvents both the use of PCR and the requirement for large amounts of initial template.
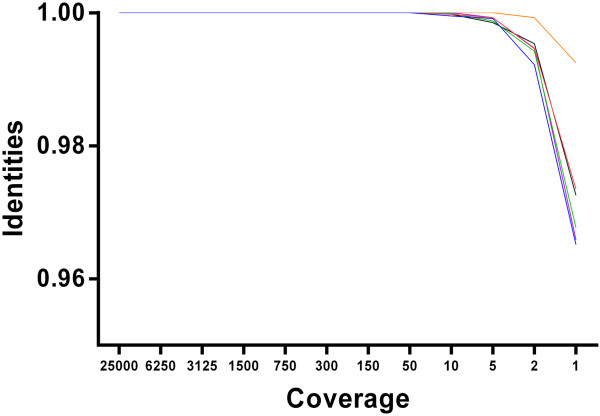
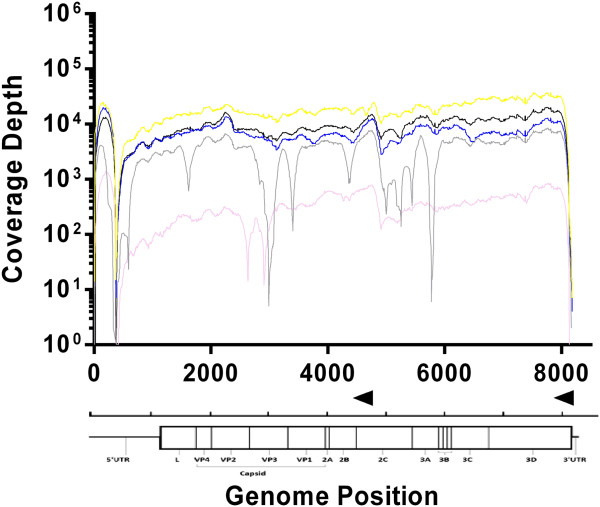
Results: The protocol was successfully validated using five FMDV positive clinical samples from the 2001 epidemic in the United Kingdom, as well as a panel of representative viruses from all seven serotypes. In addition, this protocol was successfully used to recover 94% of an FMDV genome that had previously been identified as cell culture negative. Genome sequences from three other non-FMDV polyadenylated RNA viruses (EMCV, ERAV, VESV) were also obtained with minor protocol amendments. We calculated that a minimum coverage depth of 22 reads was required to produce an accurate consensus sequence for FMDV O. This was achieved in 5 FMDV/O/UKG isolates and the type O FMDV from the serotype panel with the exception of the 5' genomic termini and area immediately flanking the poly(C) region.
Conclusions: We have developed a universal WGS method for FMDV and other polyadenylated RNA viruses. This method works successfully from a limited quantity of starting material and eliminates the requirement for genome-specific PCR amplification. This protocol has the potential to generate consensus-level sequences within a routine high-throughput diagnostic environment.
Figures
References
-
- Di Nardo A, Knowles NJ, Paton DJ. Combining livestock trade patterns with phylogenetics to help understand the spread of foot and mouth disease in sub-Saharan Africa, the Middle East and Southeast Asia. Rev Sci Tech. 2011;30(1):63–85. - PubMed
-
- Wright CF, Morelli MJ, Thebaud G, Knowles NJ, Herzyk P, Paton DJ, Haydon DT, King DP. Beyond the consensus: dissecting within-host viral population diversity of foot-and-mouth disease virus by using next-generation genome sequencing. J Virol. 2011;85(5):2266–2275. doi: 10.1128/JVI.01396-10. - DOI - PMC - PubMed
-
- Cottam EM, Wadsworth J, Shaw AE, Rowlands RJ, Goatley L, Maan S, Maan NS, Mertens PP, Ebert K, Li Y, Ryan ED, Juleff N, Ferris NP, Wilesmith JW, Haydon DT, King DP, Paton DJ, Knowles NJ. Transmission pathways of foot-and-mouth disease virus in the United Kingdom in 2007. PLoS Pathog. 2008;4(4):e1000050. doi: 10.1371/journal.ppat.1000050. - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
