

Functional roles of nucleosome stability and dynamics
- PMID: 25275099
- PMCID: PMC4366587
- DOI: 10.1093/bfgp/elu038
Functional roles of nucleosome stability and dynamics
Abstract
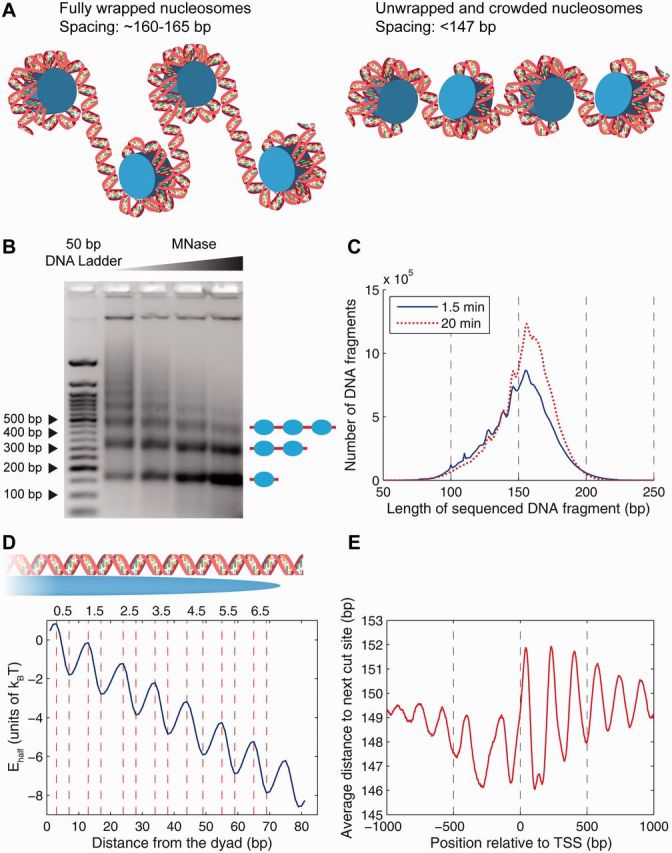
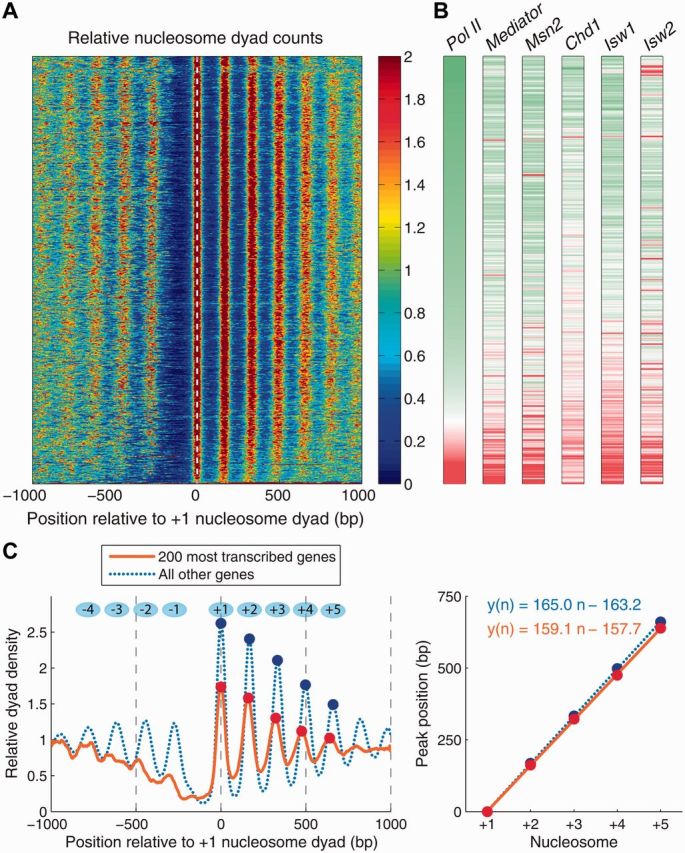
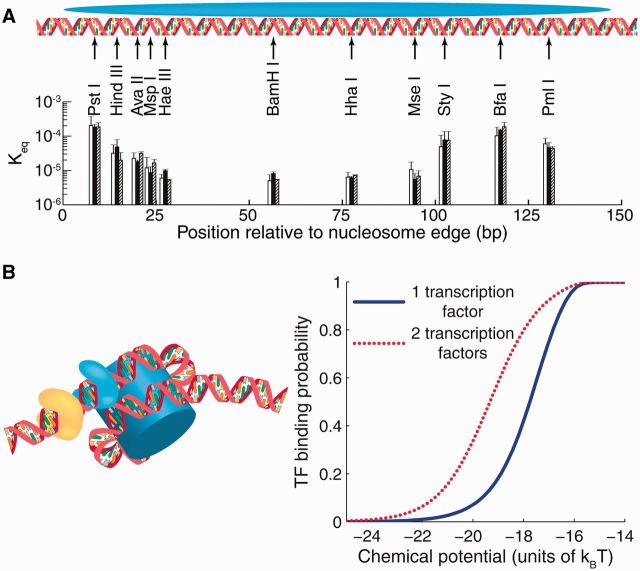
Nucleosome is a histone-DNA complex known as the fundamental repeating unit of chromatin. Up to 90% of eukaryotic DNA is wrapped around consecutive octamers made of the core histones H2A, H2B, H3 and H4. Nucleosome positioning affects numerous cellular processes that require robust and timely access to genomic DNA, which is packaged into the tight confines of the cell nucleus. In living cells, nucleosome positions are determined by intrinsic histone-DNA sequence preferences, competition between histones and other DNA-binding proteins for genomic sequence, and ATP-dependent chromatin remodelers. We discuss the major energetic contributions to nucleosome formation and remodeling, focusing especially on partial DNA unwrapping off the histone octamer surface. DNA unwrapping enables efficient access to nucleosome-buried binding sites and mediates rapid nucleosome removal through concerted action of two or more DNA-binding factors. High-resolution, genome-scale maps of distances between neighboring nucleosomes have shown that DNA unwrapping and nucleosome crowding (mutual invasion of nucleosome territories) are much more common than previously thought. Ultimately, constraints imposed by nucleosome energetics on the rates of ATP-dependent and spontaneous chromatin remodeling determine nucleosome occupancy genome-wide, and shape pathways of cellular response to environmental stresses.
Keywords: DNA; chromatin; gene regulation; nucleosome; stress response; transcription factor.
© The Author 2014. Published by Oxford University Press. All rights reserved. For permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Grosberg AY, Khokhlov AR. Statistical Physics of Macromolecules. Woodbury, NY: AIP Press; 1994.
-
- Frank-Kamenetskii MD. Biophysics of the DNA molecule. Phys Rep. 1997;288:13–60.
-
- van Holde KE. Chromatin. New York: Springer; 1989.
-
- McBryant SJ, Adams VH, Hansen JC. Chromatin architectural proteins. Chromosome Res. 2006;14:39–51. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
