

Fluorescence microscopy
- PMID: 25275114
- PMCID: PMC4711767
- DOI: 10.1101/pdb.top071795
Fluorescence microscopy
Abstract
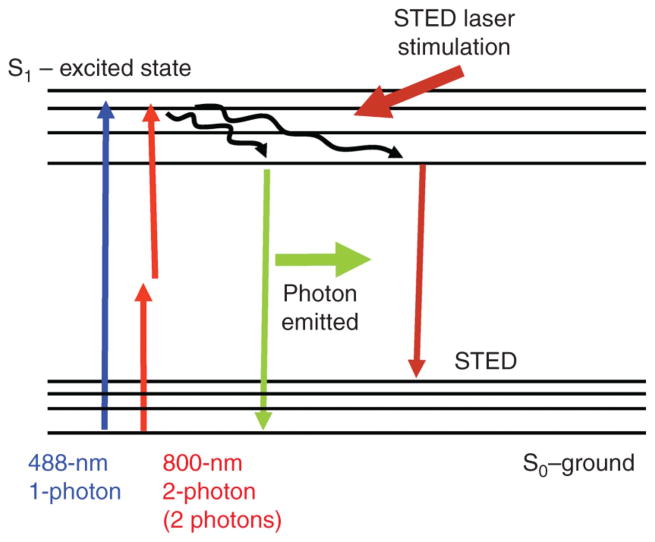
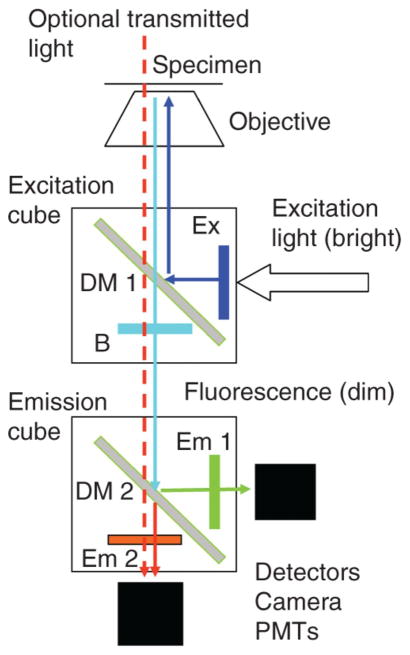
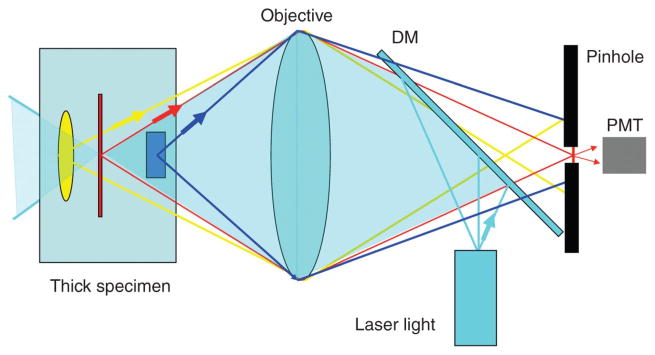
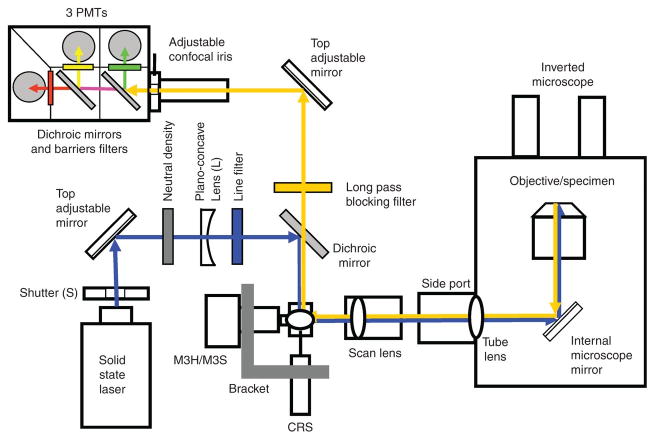
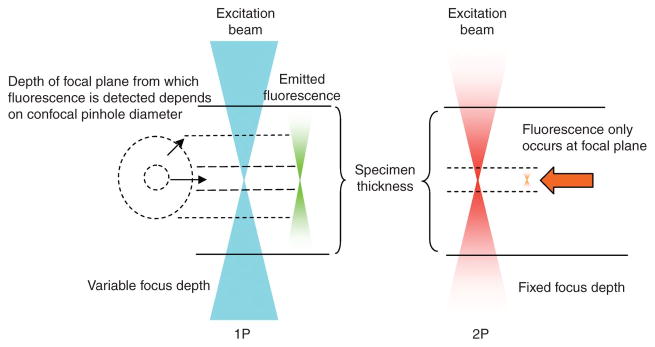
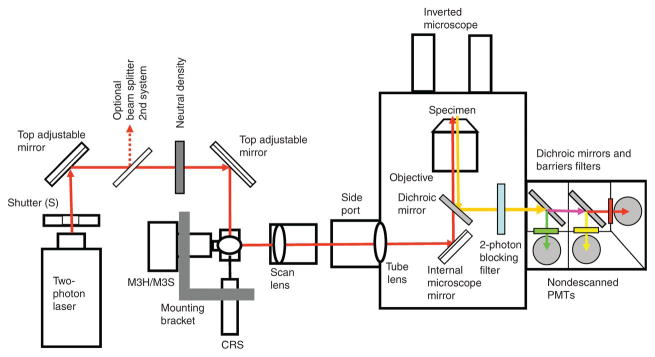
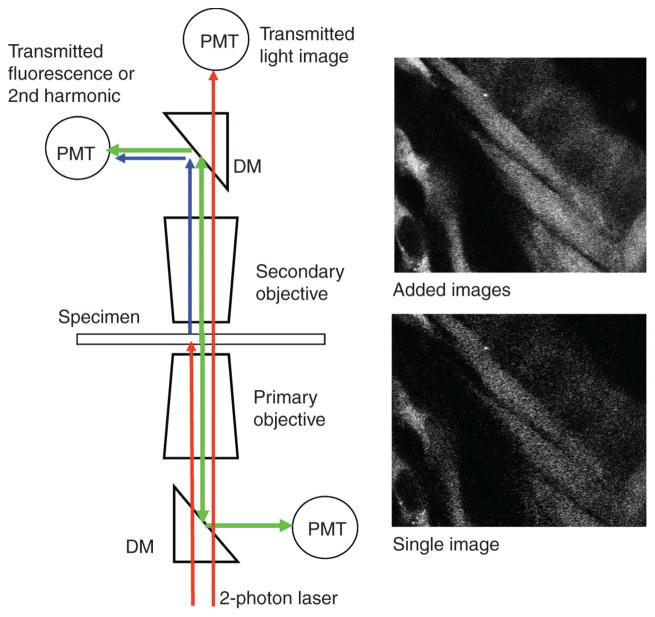
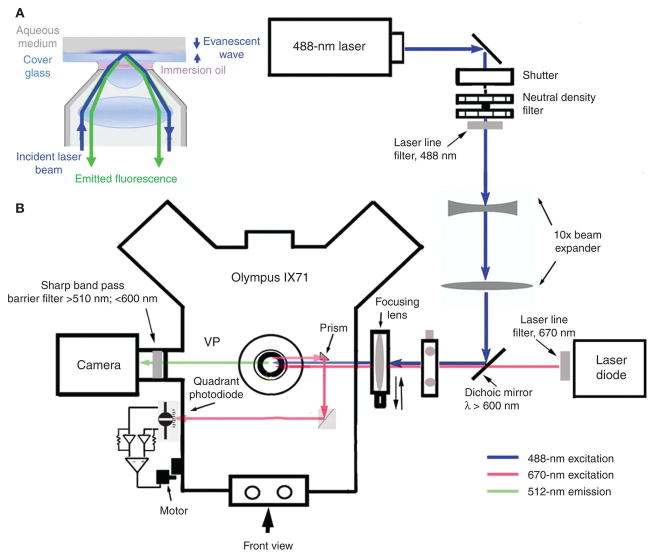
Fluorescence microscopy is a major tool with which to monitor cell physiology. Although the concepts of fluorescence and its optical separation using filters remain similar, microscope design varies with the aim of increasing image contrast and spatial resolution. The basics of wide-field microscopy are outlined to emphasize the selection, advantages, and correct use of laser scanning confocal microscopy, two-photon microscopy, scanning disk confocal microscopy, total internal reflection, and super-resolution microscopy. In addition, the principles of how these microscopes form images are reviewed to appreciate their capabilities, limitations, and constraints for operation.
© 2014 Cold Spring Harbor Laboratory Press.
Figures

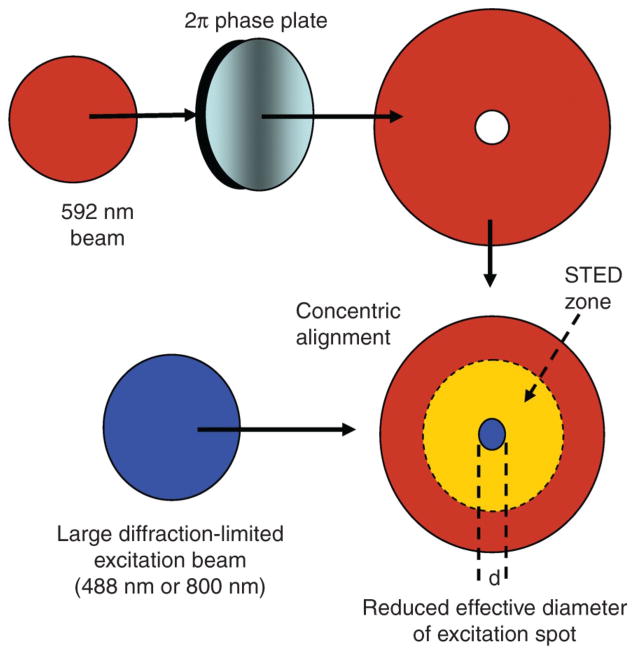
References
-
- Abbe E. Contributions to the theory of the microscope and of microscopic resolution. Arch f Mikr Anat. 1873:413–420.
-
- Ambrose EJ. A surface contact microscope for the study of cell movements. Nature. 1956;178:1194. - PubMed
-
- Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Methods Enzymol. 2003;361:1–33. - PubMed
-
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources