

Emerging technologies for the clinical microbiology laboratory
- PMID: 25278575
- PMCID: PMC4187641
- DOI: 10.1128/CMR.00003-14
Emerging technologies for the clinical microbiology laboratory
Abstract
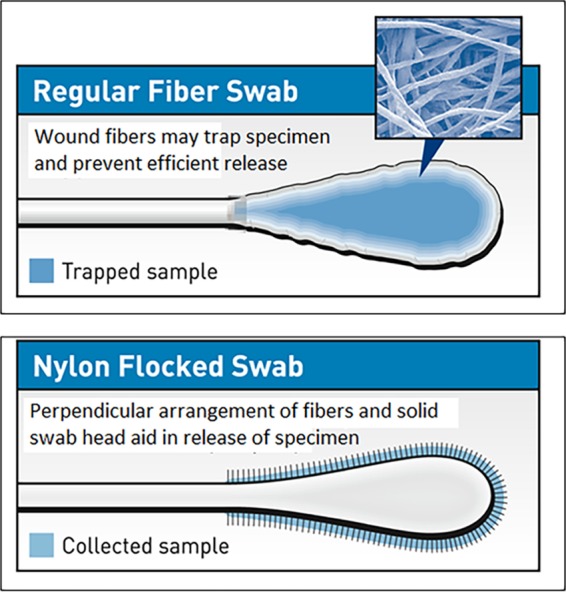
In this review we examine the literature related to emerging technologies that will help to reshape the clinical microbiology laboratory. These topics include nucleic acid amplification tests such as isothermal and point-of-care molecular diagnostics, multiplexed panels for syndromic diagnosis, digital PCR, next-generation sequencing, and automation of molecular tests. We also review matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) and electrospray ionization (ESI) mass spectrometry methods and their role in identification of microorganisms. Lastly, we review the shift to liquid-based microbiology and the integration of partial and full laboratory automation that are beginning to impact the clinical microbiology laboratory.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
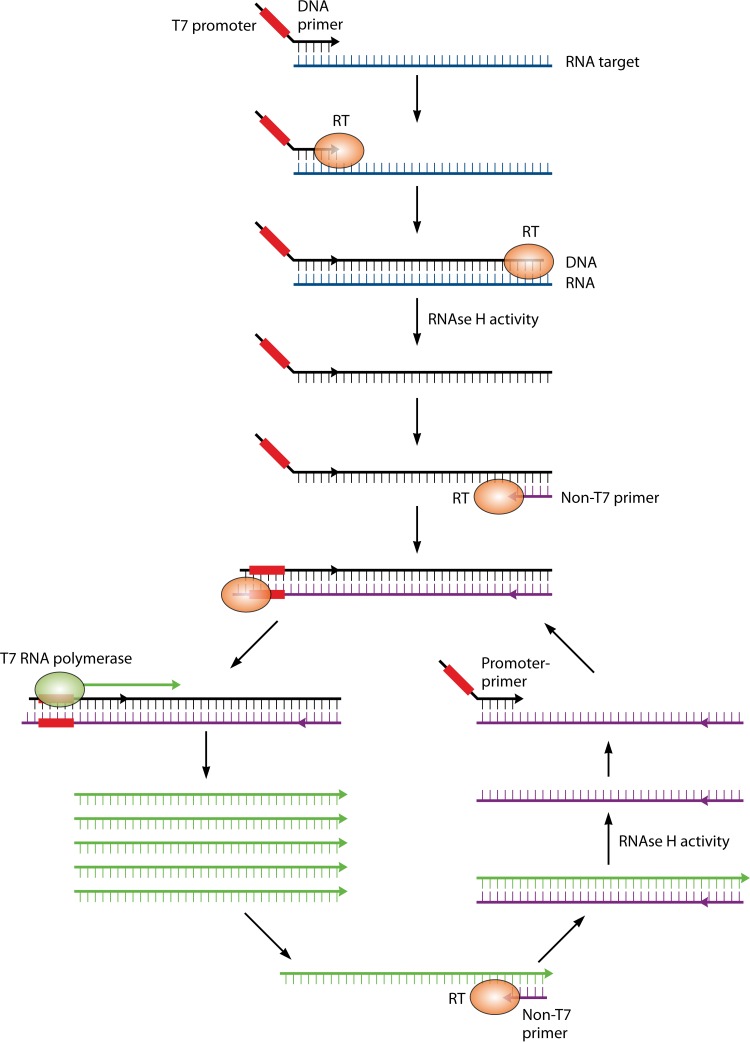
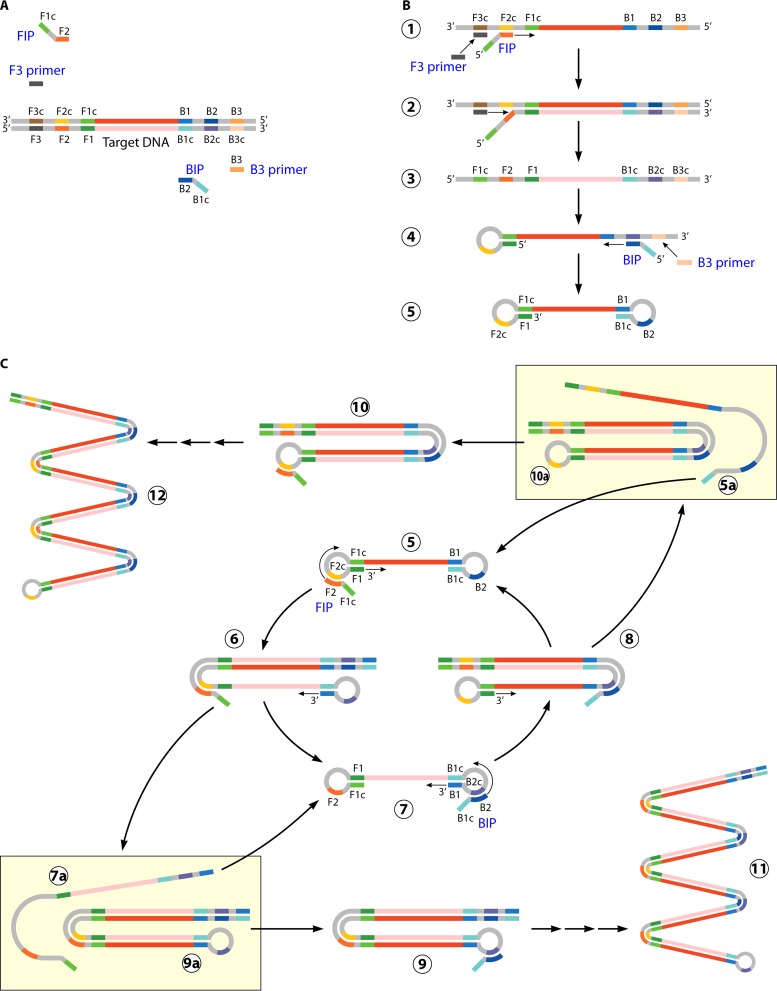
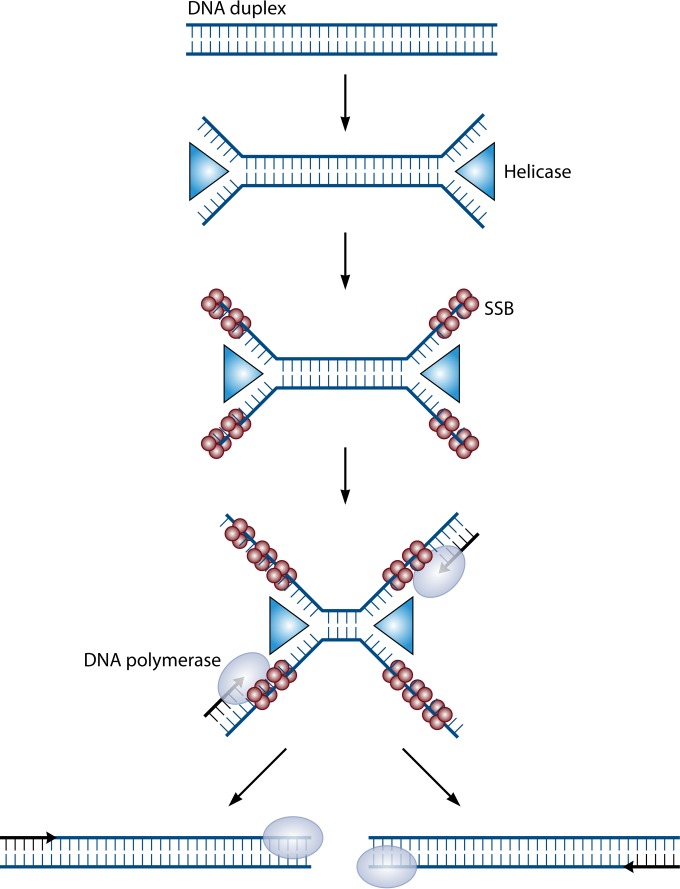
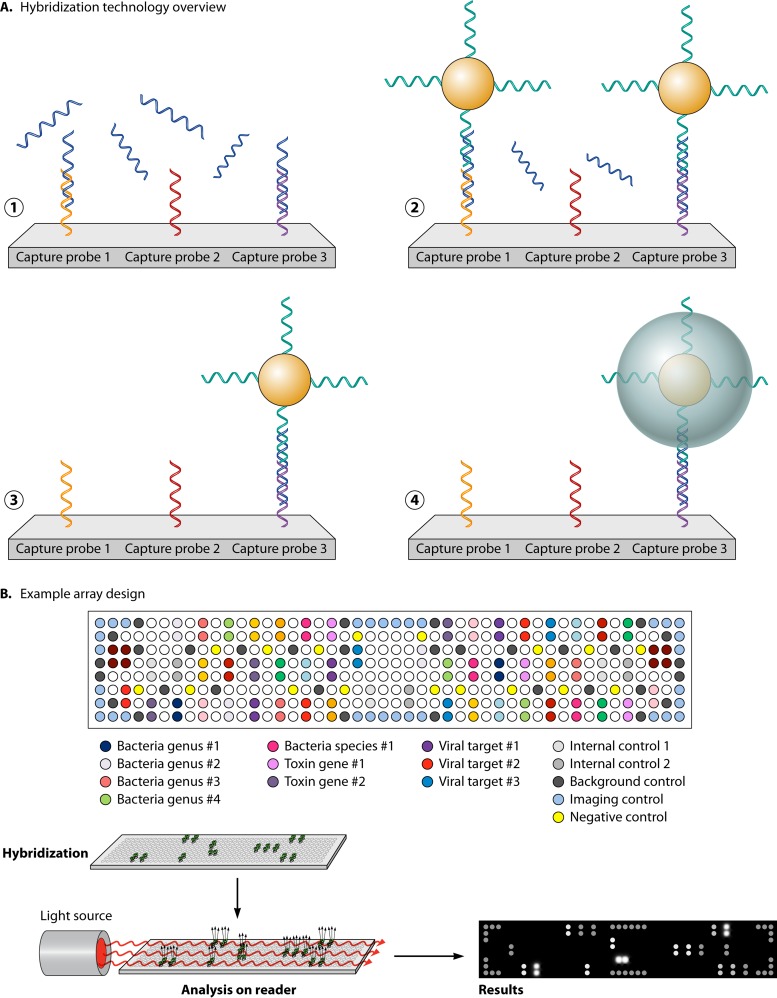
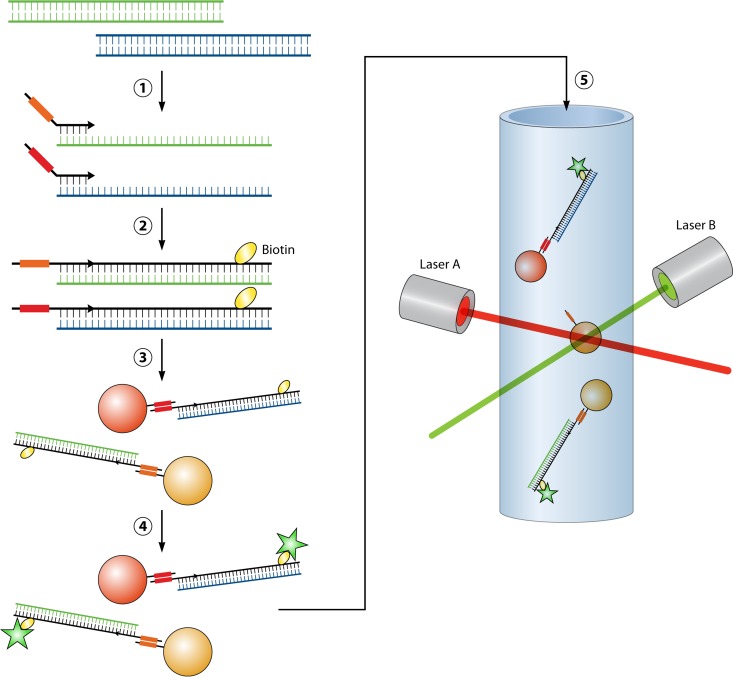
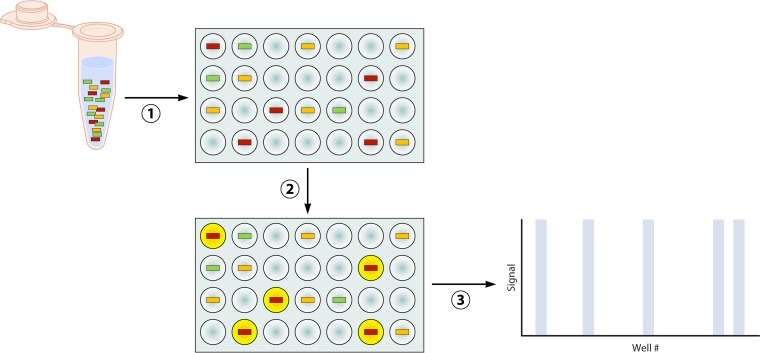
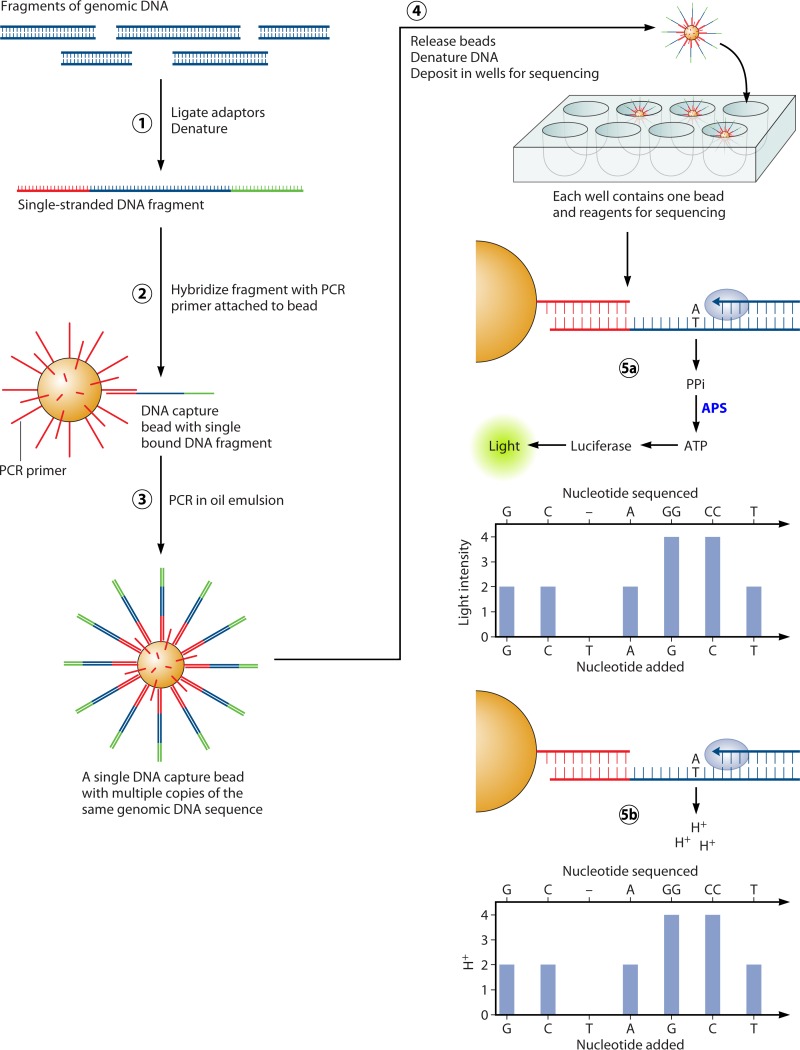
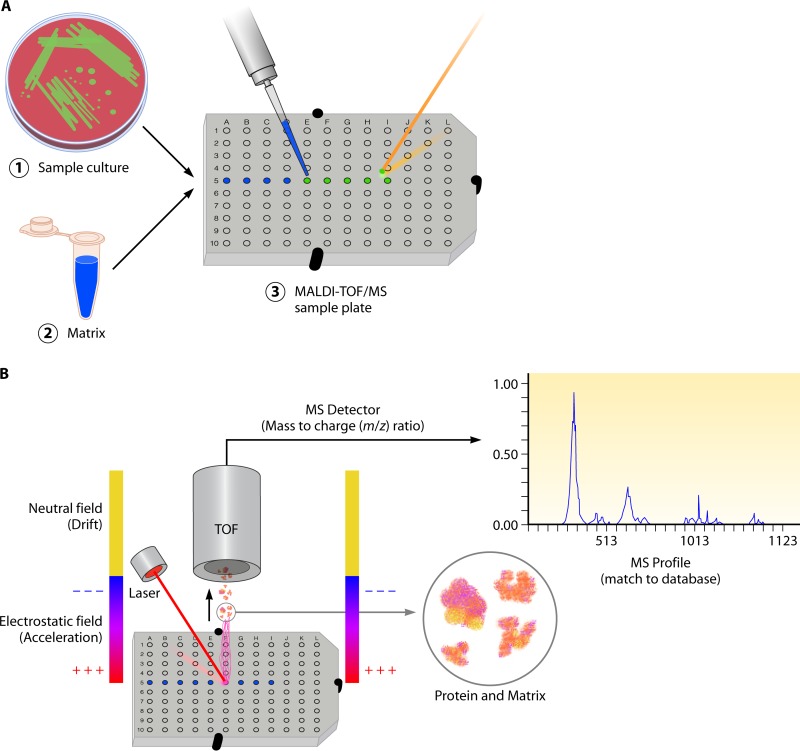




References
-
- Valenstein PN. 1990. Evaluating diagnostic tests with imperfect standards. Am. J. Clin. Pathol. 93:252–258 - PubMed
-
- CLSI. 2005. User varification of performance for precisioin and trueness. Approved guideline, 2nd ed. CLSI document EP15-A2. CLSI, Wayne, PA
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
