

Mitochondrial targets for volatile anesthetics against cardiac ischemia-reperfusion injury
- PMID: 25278902
- PMCID: PMC4165278
- DOI: 10.3389/fphys.2014.00341
Mitochondrial targets for volatile anesthetics against cardiac ischemia-reperfusion injury
Abstract
Mitochondria are critical modulators of cell function and are increasingly recognized as proximal sensors and effectors that ultimately determine the balance between cell survival and cell death. Volatile anesthetics (VA) are long known for their cardioprotective effects, as demonstrated by improved mitochondrial and cellular functions, and by reduced necrotic and apoptotic cell death during cardiac ischemia and reperfusion (IR) injury. The molecular mechanisms by which VA impart cardioprotection are still poorly understood. Because of the emerging role of mitochondria as therapeutic targets in diseases, including ischemic heart disease, it is important to know if VA-induced cytoprotective mechanisms are mediated at the mitochondrial level. In recent years, considerable evidence points to direct effects of VA on mitochondrial channel/transporter protein functions and electron transport chain (ETC) complexes as potential targets in mediating cardioprotection. This review furnishes an integrated overview of targets that VA impart on mitochondrial channels/transporters and ETC proteins that could provide a basis for cation regulation and homeostasis, mitochondrial bioenergetics, and reactive oxygen species (ROS) emission in redox signaling for cardiac cell protection during IR injury.
Keywords: cardiac IR injury; cardioprotection; electron transport chain; isoflurane; mitochondrial bioenergetics; volatile anesthetics.
Figures
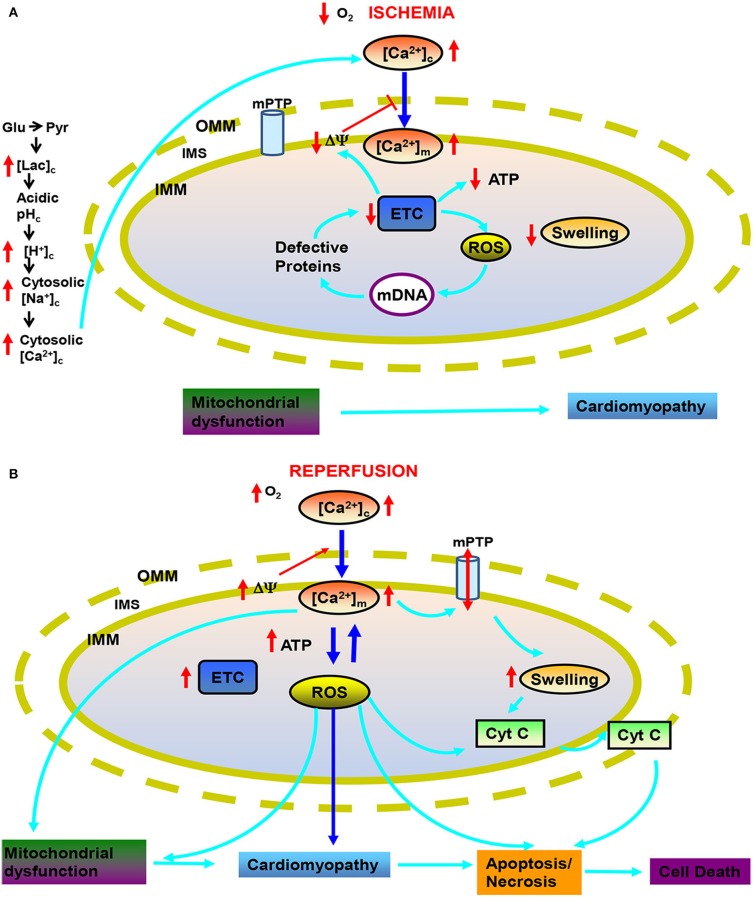
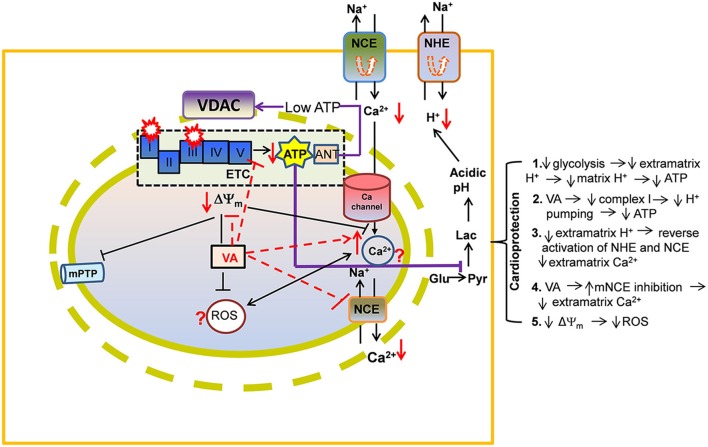
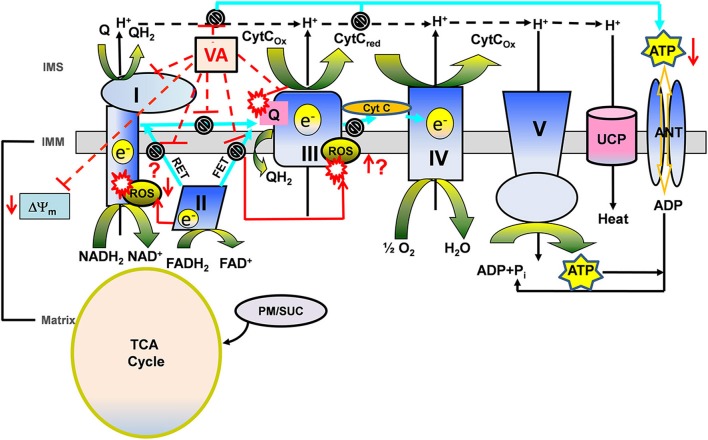
 represents reverse functioning of NHE and NCE.
represents reverse functioning of NHE and NCE.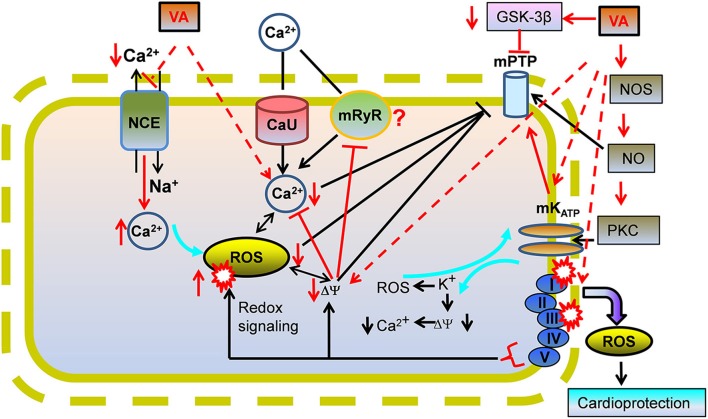
References
-
- Agarwal B., Camara A. K., Stowe D. F., Bosnjak Z. J., Dash R. K. (2012). Enhanced charge-independent mitochondrial free Ca2+ and attenuated ADP-induced NADH oxidation by isoflurane: implications for cardioprotection. Biochim. Biophys. Acta 1817, 453–465 10.1016/j.bbabio.2011.11.011 - DOI - PMC - PubMed
-
- An J., Varadarajan S. G., Novalija E., Stowe D. F. (2001). Ischemic and anesthetic preconditioning reduces cytosolic [Ca2+] and improves Ca2+ responses in intact hearts. Am. J. Physiol. Heart Circ. Physiol. 281, H1508–H1523 - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources