

Integration of heterogeneous molecular networks to unravel gene-regulation in Mycobacterium tuberculosis
- PMID: 25279447
- PMCID: PMC4181829
- DOI: 10.1186/s12918-014-0111-5
Integration of heterogeneous molecular networks to unravel gene-regulation in Mycobacterium tuberculosis
Abstract
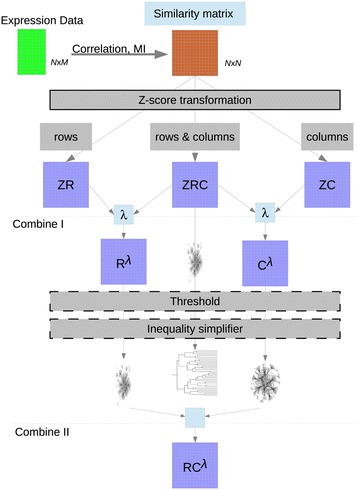
Background: Different methods have been developed to infer regulatory networks from heterogeneous omics datasets and to construct co-expression networks. Each algorithm produces different networks and efforts have been devoted to automatically integrate them into consensus sets. However each separate set has an intrinsic value that is diluted and partly lost when building a consensus network. Here we present a methodology to generate co-expression networks and, instead of a consensus network, we propose an integration framework where the different networks are kept and analysed with additional tools to efficiently combine the information extracted from each network.
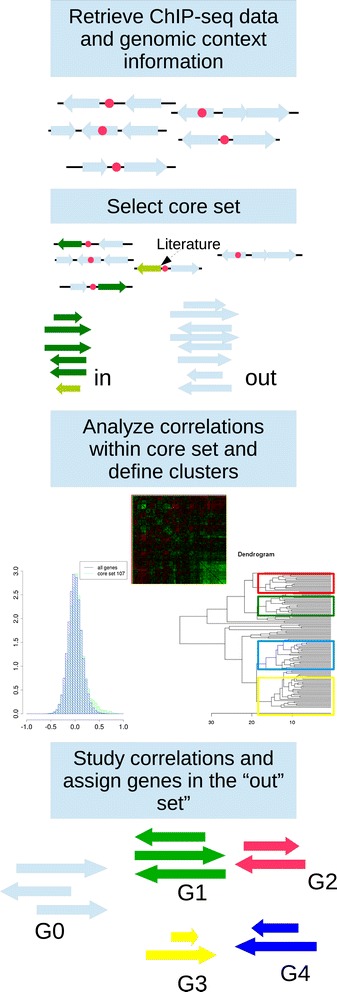
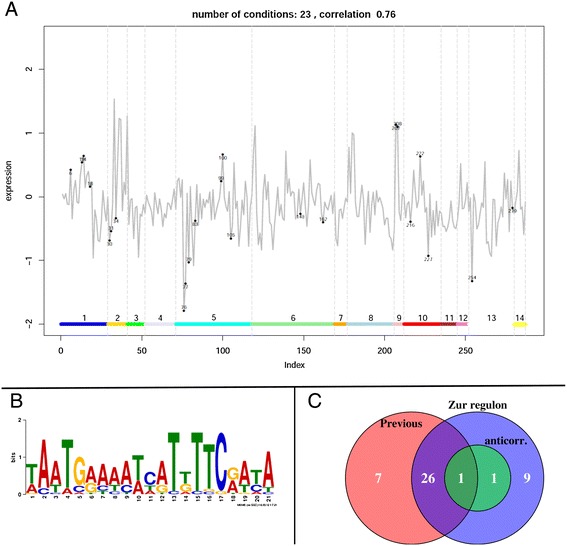
Results: We developed a workflow to efficiently analyse information generated by different inference and prediction methods. Our methodology relies on providing the user the means to simultaneously visualise and analyse the coexisting networks generated by different algorithms, heterogeneous datasets, and a suite of analysis tools. As a show case, we have analysed the gene co-expression networks of Mycobacterium tuberculosis generated using over 600 expression experiments. Regarding DNA damage repair, we identified SigC as a key control element, 12 new targets for LexA, an updated LexA binding motif, and a potential mismatch repair system. We expanded the DevR regulon with 27 genes while identifying 9 targets wrongly assigned to this regulon. We discovered 10 new genes linked to zinc uptake and a new regulatory mechanism for ZuR. The use of co-expression networks to perform system level analysis allows the development of custom made methodologies. As show cases we implemented a pipeline to integrate ChIP-seq data and another method to uncover multiple regulatory layers.
Conclusions: Our workflow is based on representing the multiple types of information as network representations and presenting these networks in a synchronous framework that allows their simultaneous visualization while keeping specific associations from the different networks. By simultaneously exploring these networks and metadata, we gained insights into regulatory mechanisms in M. tuberculosis that could not be obtained through the separate analysis of each data type.
Figures





References
-
- De Smet R, Marchal K. Advantages and limitations of current network inference methods. Nat Rev Micro. 2010;8:717–729. - PubMed
-
- Cantone I, Marucci L, Iorio F, Ricci MA, Belcastro V, Bansal M, Santini S, di Bernardo M, di Bernardo D, Cosma MP. A yeast synthetic network for in vivo assessment of reverse-engineering and modeling approaches. Cell. 2009;137:172–181. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
