

Cryptic and complex chromosomal aberrations in early-onset neuropsychiatric disorders
- PMID: 25279985
- PMCID: PMC4185111
- DOI: 10.1016/j.ajhg.2014.09.005
Cryptic and complex chromosomal aberrations in early-onset neuropsychiatric disorders
Abstract
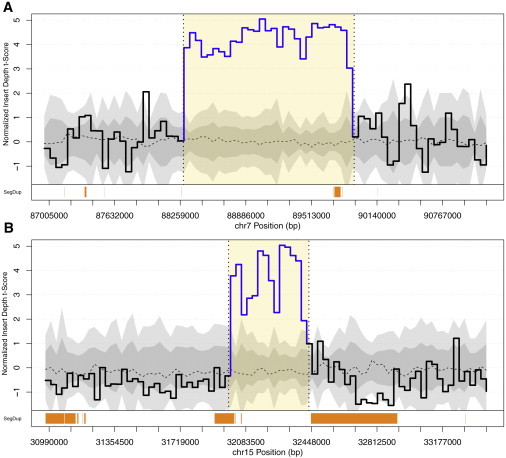
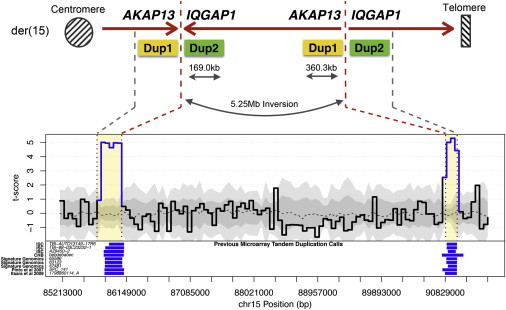
Structural variation (SV) is a significant component of the genetic etiology of both neurodevelopmental and psychiatric disorders; however, routine guidelines for clinical genetic screening have been established only in the former category. Genome-wide chromosomal microarray (CMA) can detect genomic imbalances such as copy-number variants (CNVs), but balanced chromosomal abnormalities (BCAs) still require karyotyping for clinical detection. Moreover, submicroscopic BCAs and subarray threshold CNVs are intractable, or cryptic, to both CMA and karyotyping. Here, we performed whole-genome sequencing using large-insert jumping libraries to delineate both cytogenetically visible and cryptic SVs in a single test among 30 clinically referred youth representing a range of severe neuropsychiatric conditions. We detected 96 SVs per person on average that passed filtering criteria above our highest-confidence resolution (6,305 bp) and an additional 111 SVs per genome below this resolution. These SVs rearranged 3.8 Mb of genomic sequence and resulted in 42 putative loss-of-function (LoF) or gain-of-function mutations per person. We estimate that 80% of the LoF variants were cryptic to clinical CMA. We found myriad complex and cryptic rearrangements, including a "paired" duplication (360 kb, 169 kb) that flanks a 5.25 Mb inversion that appears in 7 additional cases from clinical CNV data among 47,562 individuals. Following convergent genomic profiling of these independent clinical CNV data, we interpreted three SVs to be of potential clinical significance. These data indicate that sequence-based delineation of the full SV mutational spectrum warrants exploration in youth referred for neuropsychiatric evaluation and clinical diagnostic SV screening more broadly.
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
