

Mutation rates and evolution of multiple coding in RNA-based protocells
- PMID: 25280530
- PMCID: PMC4247474
- DOI: 10.1007/s00239-014-9648-6
Mutation rates and evolution of multiple coding in RNA-based protocells
Abstract
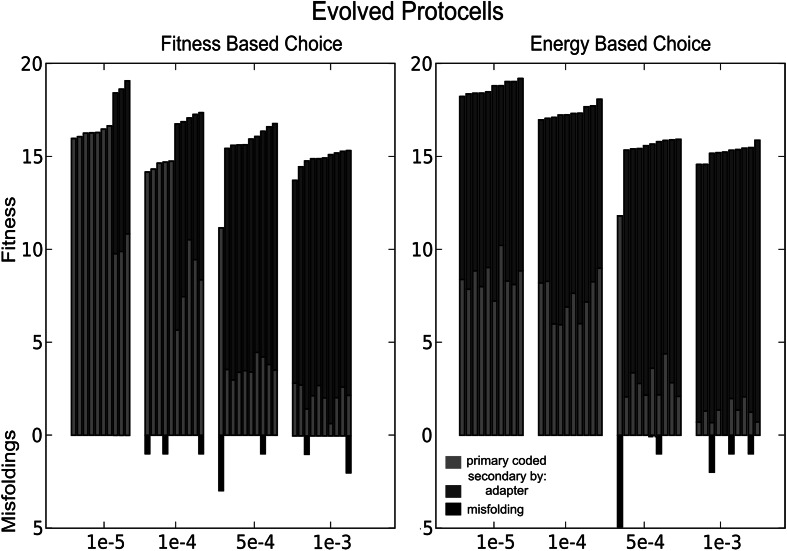
RNA has a myriad of biological roles in contemporary life. We use the RNA paradigm for genotype-phenotype mappings to study the evolution of multiple coding in dependence to mutation rates. We study three different one-to-many genotype-phenotype mappings which have the potential to encode the information for multiple functions on a single sequence. These three different maps are (i) cofolding, where two sequences can bind and "cofold," (ii) suboptimal folding, where the alternative foldings within a certain range of the native state of sequences are considered, and (iii) adapter-based folding, in which protocells can evolve adapter-mediated alternative foldings. We study how protocells with a set of sequences can code for a set of predefined functional structures, while avoiding all other structures, which are considered to be misfoldings. Note that such misfolded structures are far more prevalent than functional ones. Our results highlight the flexibility of the RNA sequence to secondary structure mapping and the power of evolution to shape the genotype-phenotype mapping. We show that high fitness can be achieved even at high mutation rates. Mutation rates affect genome size, but differently depending on which folding method is used. We observe that cofolding limits the possibility to avoid misfolded structures and that adapters are always beneficial for fitness, but even more beneficial at low mutation rates. In all cases, the evolution procedure selects for molecules that can form additional structures. Our results indicate that inherent properties of RNA molecules and their interactions allow the evolution of complexity even at high mutation rates.
Figures






References
-
- Attolini CS-O, Stadler PF. Neutral networks of interacting RNA secondary structures. Adv Complex Syst. 2005;08(02n03):275–283. doi: 10.1142/S0219525905000427. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
