

CanvasDB: a local database infrastructure for analysis of targeted- and whole genome re-sequencing projects
- PMID: 25281234
- PMCID: PMC4184106
- DOI: 10.1093/database/bau098
CanvasDB: a local database infrastructure for analysis of targeted- and whole genome re-sequencing projects
Abstract
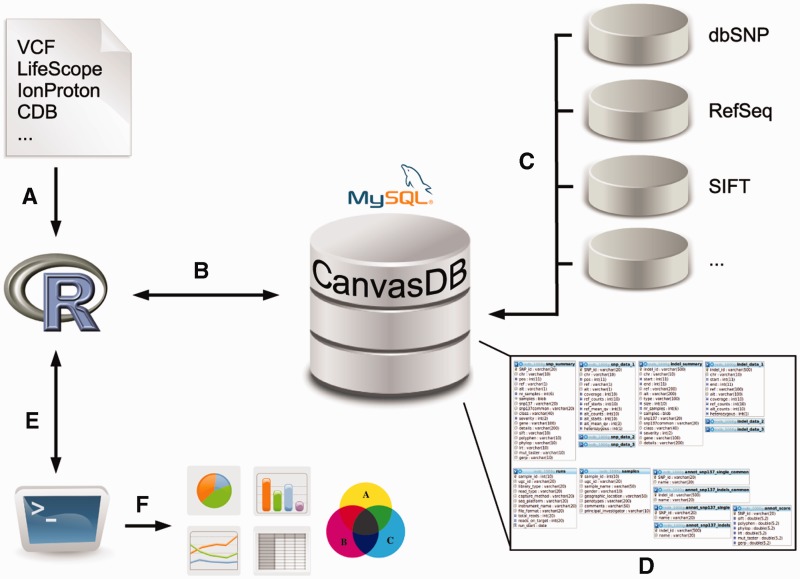
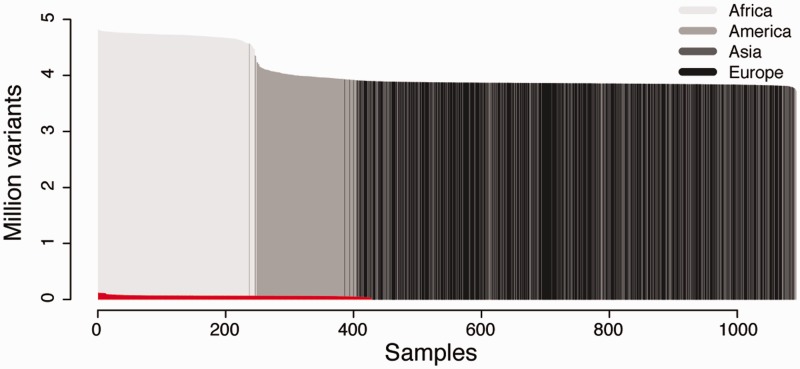
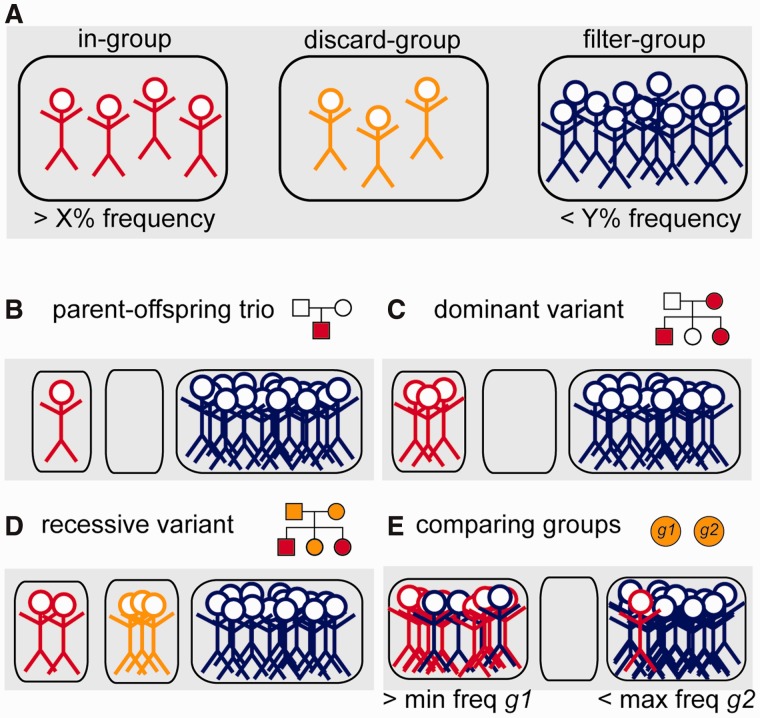
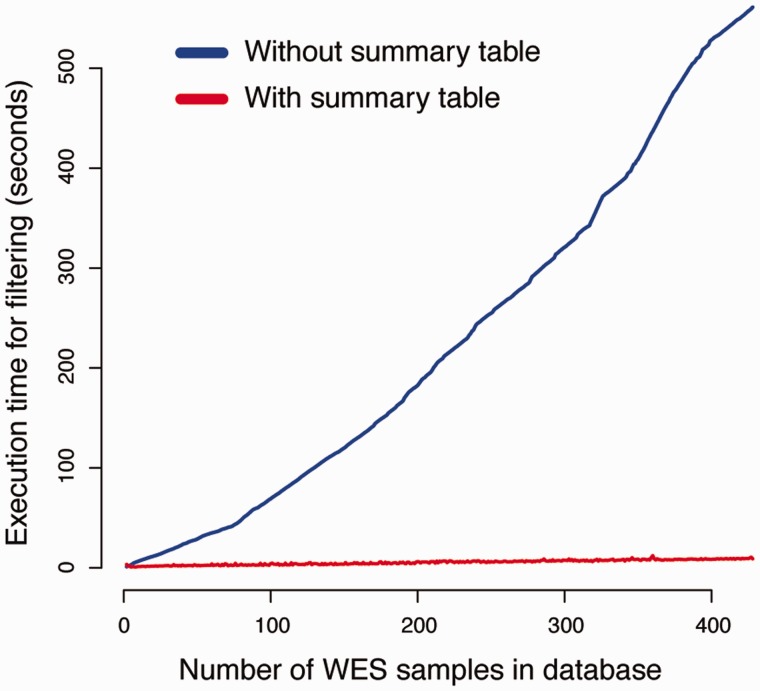
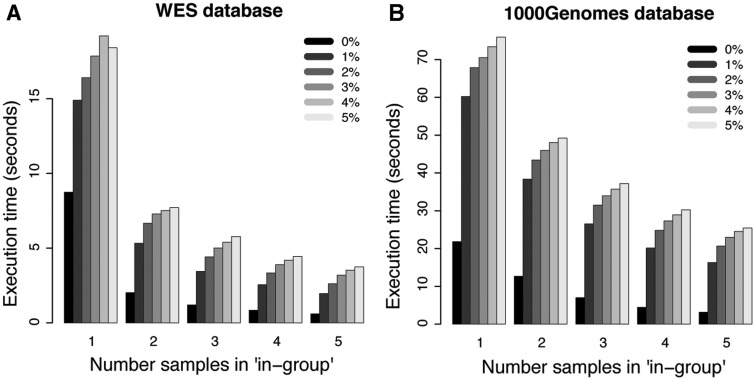
CanvasDB is an infrastructure for management and analysis of genetic variants from massively parallel sequencing (MPS) projects. The system stores SNP and indel calls in a local database, designed to handle very large datasets, to allow for rapid analysis using simple commands in R. Functional annotations are included in the system, making it suitable for direct identification of disease-causing mutations in human exome- (WES) or whole-genome sequencing (WGS) projects. The system has a built-in filtering function implemented to simultaneously take into account variant calls from all individual samples. This enables advanced comparative analysis of variant distribution between groups of samples, including detection of candidate causative mutations within family structures and genome-wide association by sequencing. In most cases, these analyses are executed within just a matter of seconds, even when there are several hundreds of samples and millions of variants in the database. We demonstrate the scalability of canvasDB by importing the individual variant calls from all 1092 individuals present in the 1000 Genomes Project into the system, over 4.4 billion SNPs and indels in total. Our results show that canvasDB makes it possible to perform advanced analyses of large-scale WGS projects on a local server. Database URL: https://github.com/UppsalaGenomeCenter/CanvasDB.
© The Author(s) 2014. Published by Oxford University Press.
Figures
References
-
- de Ligt J., Willemsen M.H., van Bon B.W., et al. (2012) Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med., 367, 1921–1929 - PubMed
-
- Rauch A., Wieczorek D., Graf E., et al. (2012) Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet, 380, 1674–1682 - PubMed
-
- Li Y., Vinckenbosch N., Tian G., et al. (2010) Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet., 42, 969–972 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
