

P-REX1 creates a positive feedback loop to activate growth factor receptor, PI3K/AKT and MEK/ERK signaling in breast cancer
- PMID: 25284585
- PMCID: PMC4387124
- DOI: 10.1038/onc.2014.328
P-REX1 creates a positive feedback loop to activate growth factor receptor, PI3K/AKT and MEK/ERK signaling in breast cancer
Abstract
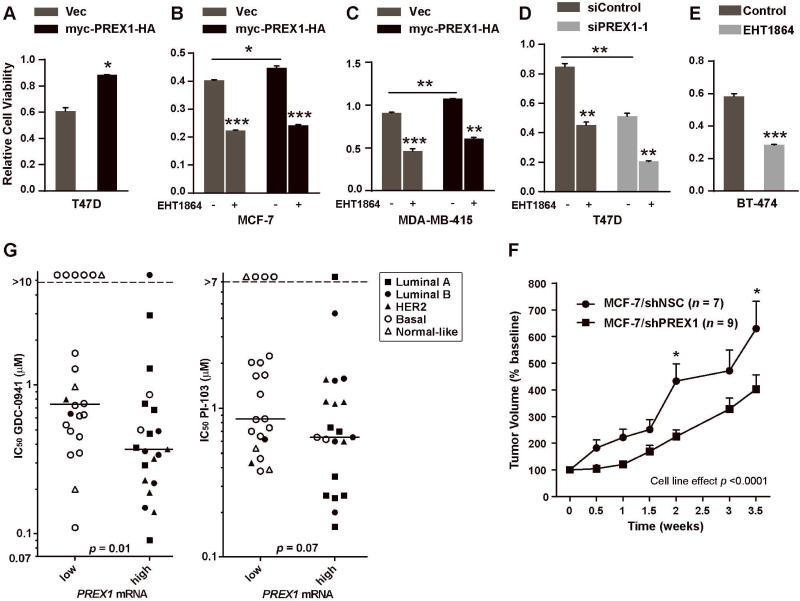
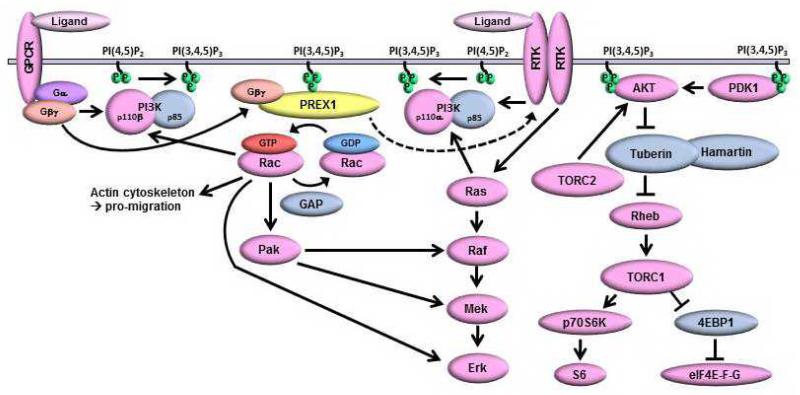
Phosphatidylinositol 3-kinase (PI3K) promotes cancer cell survival, migration, growth and proliferation by generating phosphatidylinositol 3,4,5-trisphosphate (PIP3) in the inner leaflet of the plasma membrane. PIP3 recruits pleckstrin homology domain-containing proteins to the membrane to activate oncogenic signaling cascades. Anticancer therapeutics targeting the PI3K/AKT/mTOR (mammalian target of rapamycin) pathway are in clinical development. In a mass spectrometric screen to identify PIP3-regulated proteins in breast cancer cells, levels of the Rac activator PIP3-dependent Rac exchange factor-1 (P-REX1) increased in response to PI3K inhibition, and decreased upon loss of the PI3K antagonist phosphatase and tensin homolog (PTEN). P-REX1 mRNA and protein levels were positively correlated with ER expression, and inversely correlated with PI3K pathway activation in breast tumors as assessed by gene expression and phosphoproteomic analyses. P-REX1 increased activation of Rac1, PI3K/AKT and MEK/ERK signaling in a PTEN-independent manner, and promoted cell and tumor viability. Loss of P-REX1 or inhibition of Rac suppressed PI3K/AKT and MEK/ERK, and decreased viability. P-REX1 also promoted insulin-like growth factor-1 receptor activation, suggesting that P-REX1 provides positive feedback to activators upstream of PI3K. In support of a model where PIP3-driven P-REX1 promotes both PI3K/AKT and MEK/ERK signaling, high levels of P-REX1 mRNA (but not phospho-AKT or a transcriptomic signature of PI3K activation) were predictive of sensitivity to PI3K inhibitors among breast cancer cell lines. P-REX1 expression was highest in estrogen receptor-positive breast tumors compared with many other cancer subtypes, suggesting that neutralizing the P-REX1/Rac axis may provide a novel therapeutic approach to selectively abrogate oncogenic signaling in breast cancer cells.
Figures
References
-
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–62. - PubMed
-
- Whitman M, Downes CP, Keeler M, Keller T, Cantley L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature. 1988;332(6165):644–6. - PubMed
-
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. Journal of Biological Chemistry. 1998;273(22):13375–13378. - PubMed
-
- Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11(2):317–28. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
