

Role of the ubiquitin-proteasome system in cardiac dysfunction of adipose triglyceride lipase-deficient mice
- PMID: 25285770
- PMCID: PMC4263609
- DOI: 10.1016/j.yjmcc.2014.09.028
Role of the ubiquitin-proteasome system in cardiac dysfunction of adipose triglyceride lipase-deficient mice
Abstract
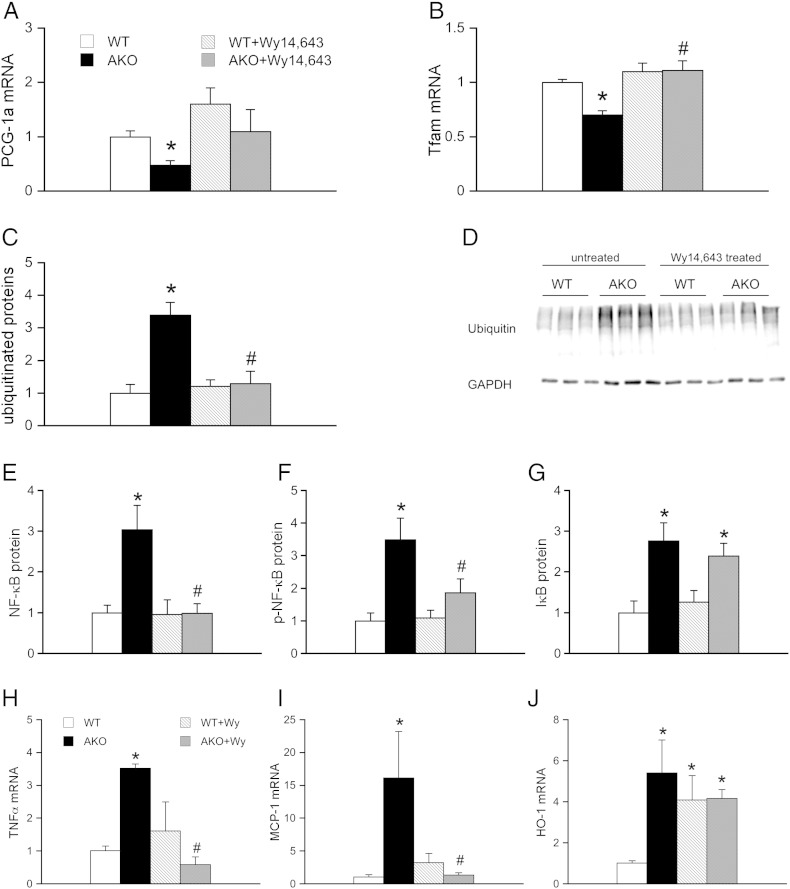
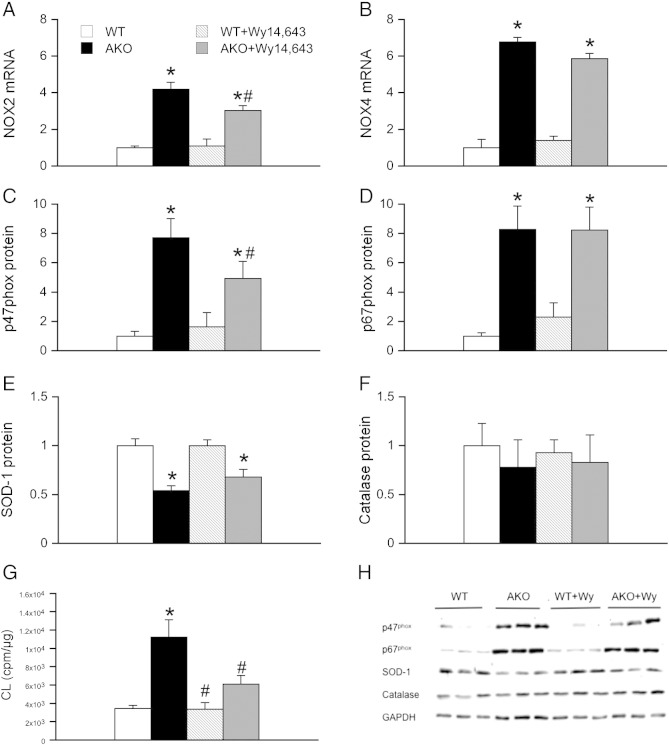
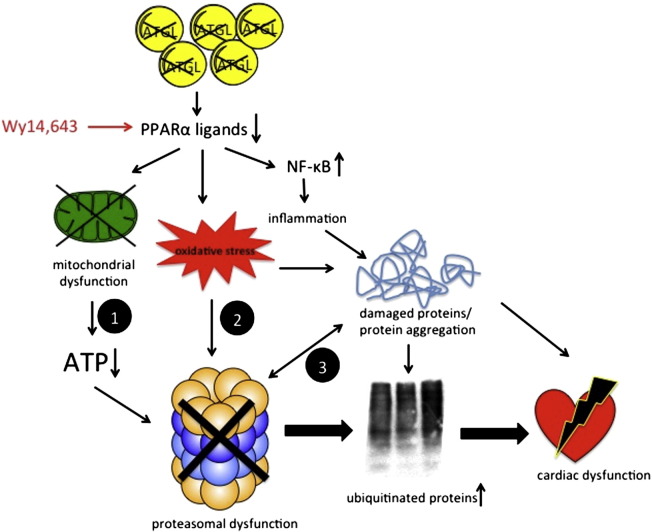
Systemic deletion of the gene encoding for adipose triglyceride lipase (ATGL) in mice leads to severe cardiac dysfunction due to massive accumulation of neutral lipids in cardiomyocytes. Recently, impaired peroxisome proliferator-activated receptor α (PPARα) signaling has been described to substantially contribute to the observed cardiac phenotype. Disturbances of the ubiquitin-proteasome system (UPS) have been implicated in numerous cardiac diseases including cardiomyopathy, ischemic heart disease, and heart failure. The objective of the present study was to investigate the potential role of UPS in cardiac ATGL deficiency. Our results demonstrate prominent accumulation of ubiquitinated proteins in hearts of ATGL-deficient mice, an effect that was abolished upon cardiomyocyte-directed overexpression of ATGL. In parallel, cardiac protein expression of the ubiquitin-activating enzyme E1a, which catalyzes the first step of the ubiquitination cascade, was significantly upregulated in ATGL-deficient hearts. Dysfunction of the UPS was accompanied by activation of NF-κB signaling. Moreover, the endoplasmic reticulum (ER)-resident chaperon protein disulfide isomerase was significantly upregulated in ATGL-deficient hearts. Chronic treatment of ATGL-deficient mice with the PPARα agonist Wy14,643 improved proteasomal function, prevented NF-κB activation and decreased oxidative stress. In summary, our data point to a hitherto unrecognized link between proteasomal function, PPARα signaling and cardiovascular disease.
Keywords: Adipose triglyceride lipase; Cardiac dysfunction; Inflammation; NF-κB; Peroxisome proliferator-activated receptor α.
Copyright © 2014. Published by Elsevier Ltd.
Figures


Similar articles
-
Cardiac dysfunction in adipose triglyceride lipase deficiency: treatment with a PPARα agonist.Br J Pharmacol. 2012 Jan;165(2):380-9. doi: 10.1111/j.1476-5381.2011.01490.x. Br J Pharmacol. 2012. PMID: 21585347 Free PMC article.
-
Cardiac oxidative stress in a mouse model of neutral lipid storage disease.Biochim Biophys Acta. 2013 Nov;1831(11):1600-8. doi: 10.1016/j.bbalip.2013.07.004. Epub 2013 Jul 15. Biochim Biophys Acta. 2013. PMID: 23867907 Free PMC article.
-
Early structural and metabolic cardiac remodelling in response to inducible adipose triglyceride lipase ablation.Cardiovasc Res. 2013 Aug 1;99(3):442-51. doi: 10.1093/cvr/cvt124. Epub 2013 May 25. Cardiovasc Res. 2013. PMID: 23708736 Free PMC article.
-
The role of adipose triglyceride lipase in lipid and glucose homeostasis: lessons from transgenic mice.Lipids Health Dis. 2019 Nov 22;18(1):204. doi: 10.1186/s12944-019-1151-z. Lipids Health Dis. 2019. PMID: 31757217 Free PMC article. Review.
-
Role of the ubiquitin-proteasome system in brain ischemia: friend or foe?Prog Neurobiol. 2014 Jan;112:50-69. doi: 10.1016/j.pneurobio.2013.10.003. Epub 2013 Oct 22. Prog Neurobiol. 2014. PMID: 24157661 Review.
Cited by
-
Regression from pathological hypertrophy in mice is sexually dimorphic and stimulus specific.Am J Physiol Heart Circ Physiol. 2022 May 1;322(5):H785-H797. doi: 10.1152/ajpheart.00644.2021. Epub 2022 Mar 18. Am J Physiol Heart Circ Physiol. 2022. PMID: 35302880 Free PMC article.
-
Curcumin may induce lipolysis via proteo-stress in Huh7 human hepatoma cells.J Clin Biochem Nutr. 2019 Sep;65(2):91-98. doi: 10.3164/jcbn.19-7. Epub 2019 Sep 1. J Clin Biochem Nutr. 2019. PMID: 31592057 Free PMC article.
-
Bcl-2 inhibition combined with PPARα activation synergistically targets leukemic stem cell-like cells in acute myeloid leukemia.Cell Death Dis. 2023 Aug 29;14(8):573. doi: 10.1038/s41419-023-06075-6. Cell Death Dis. 2023. PMID: 37644011 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
