

Loss of p53 induces cell proliferation via Ras-independent activation of the Raf/Mek/Erk signaling pathway
- PMID: 25288756
- PMCID: PMC4210339
- DOI: 10.1073/pnas.1417549111
Loss of p53 induces cell proliferation via Ras-independent activation of the Raf/Mek/Erk signaling pathway
Abstract
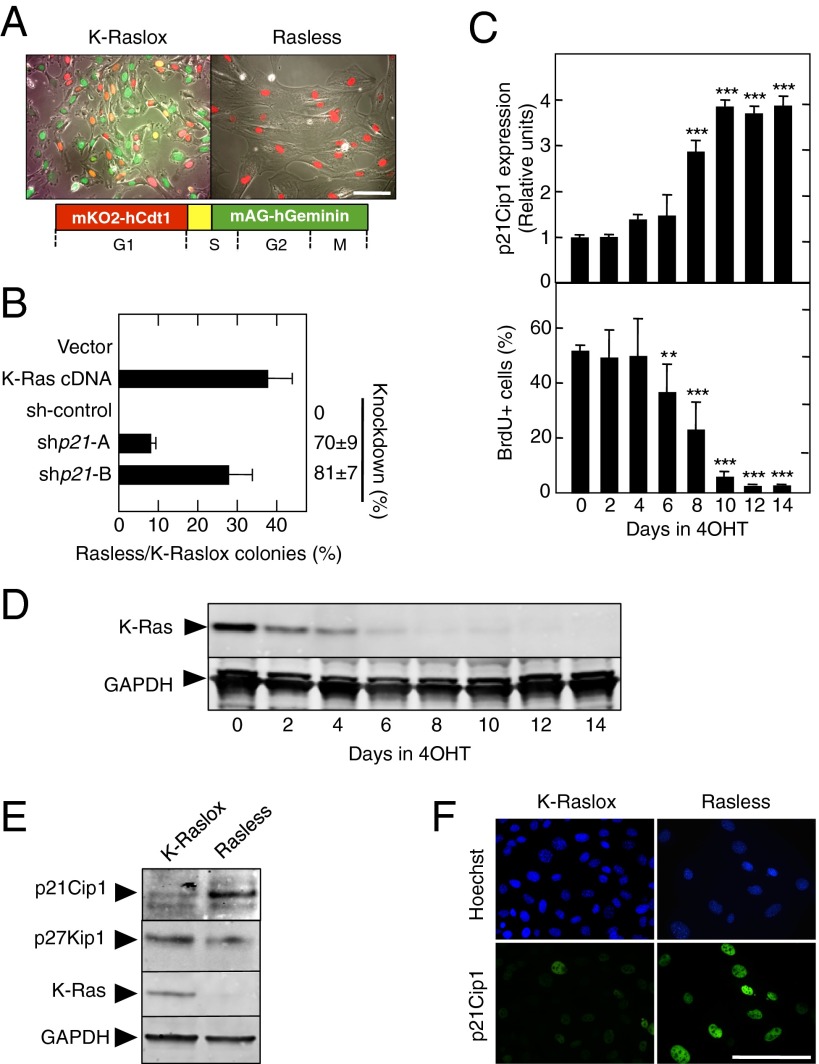
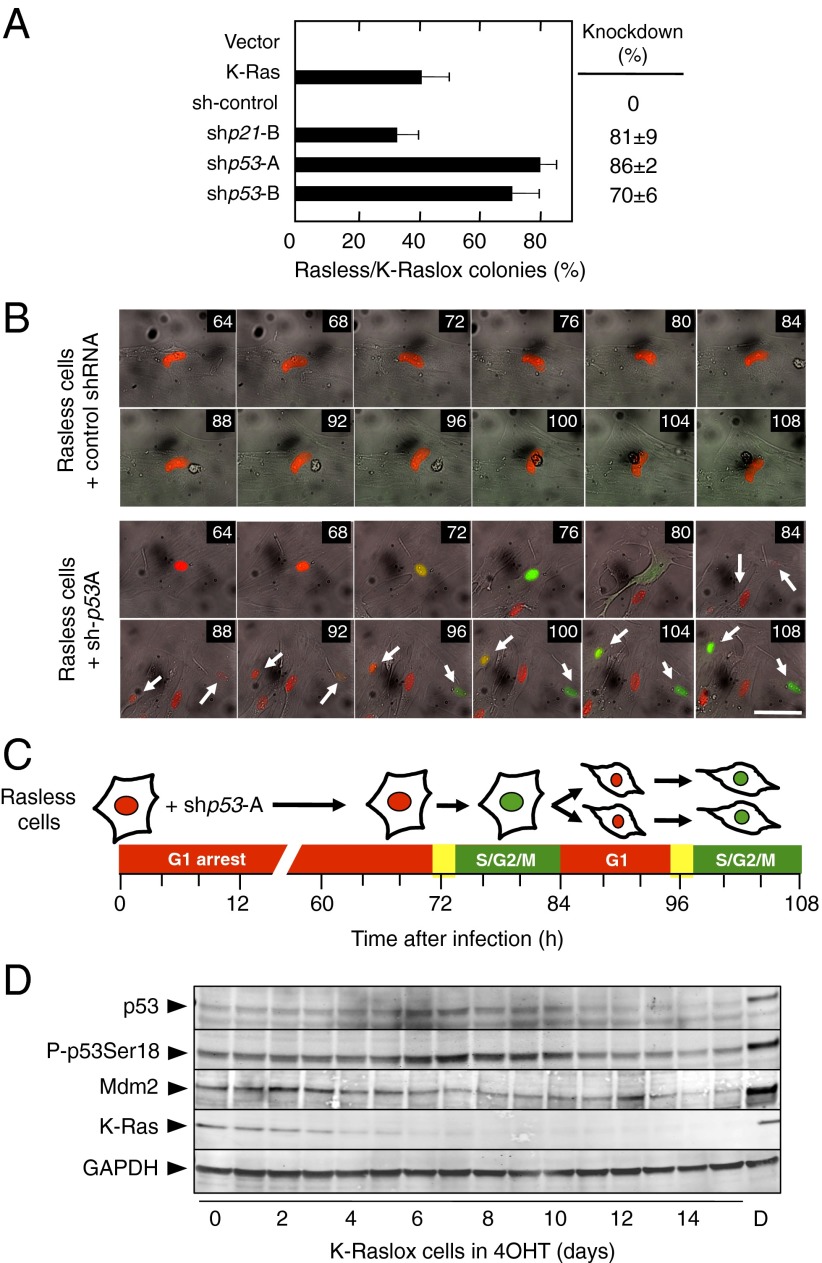
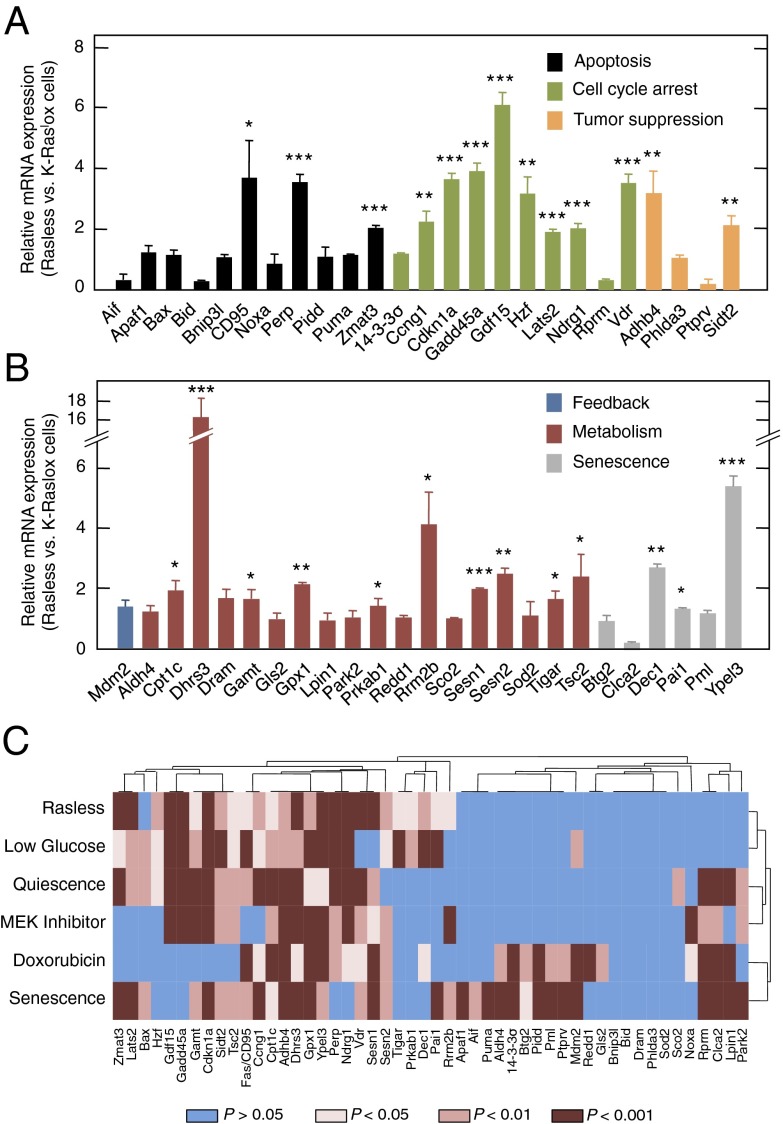
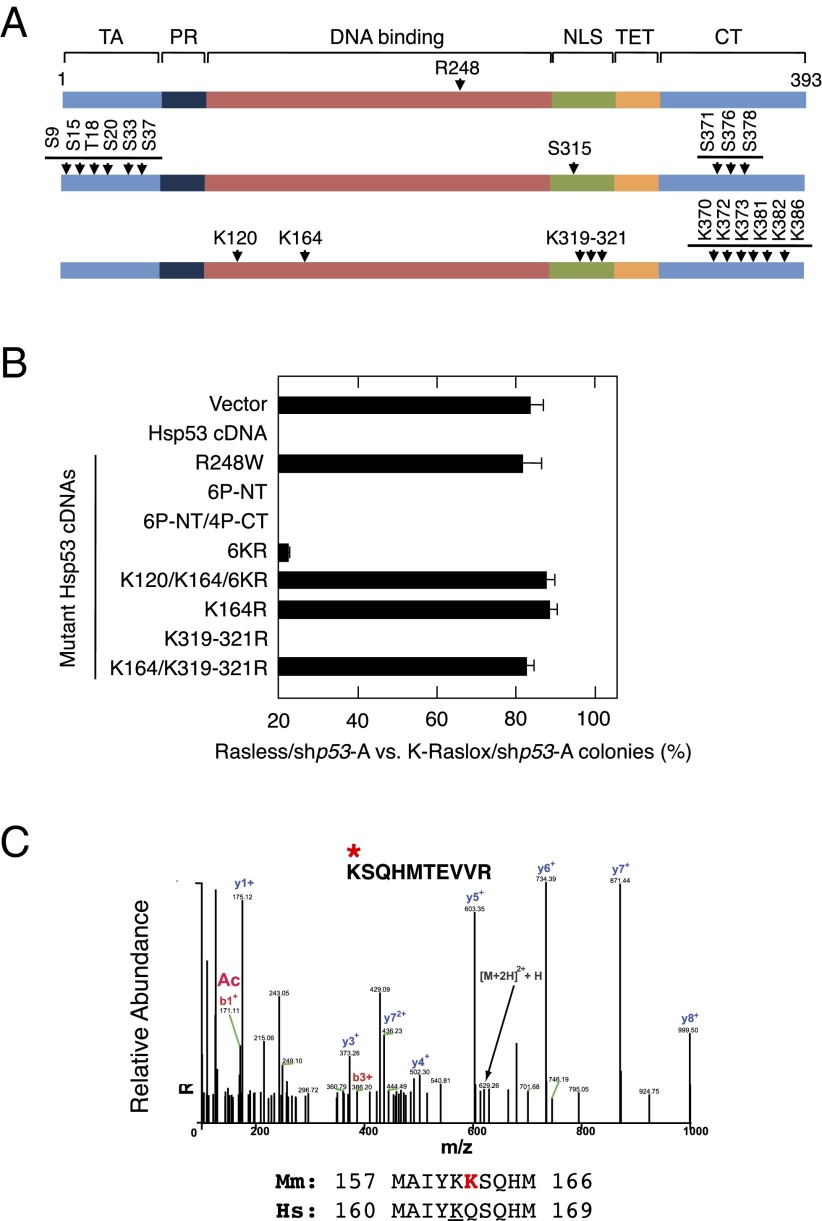
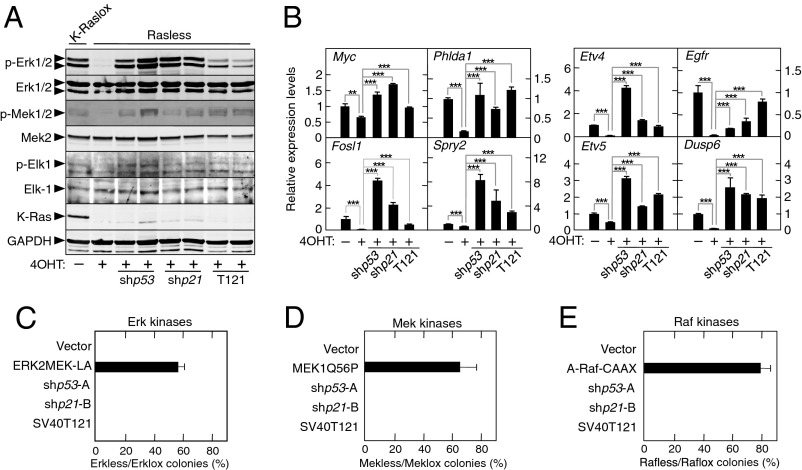
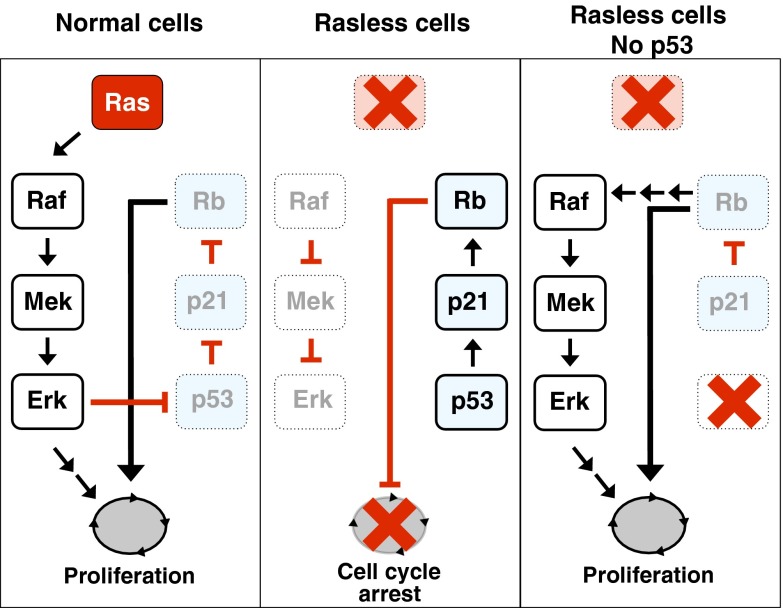
The Ras family of small GTPases constitutes a central node in the transmission of mitogenic stimuli to the cell cycle machinery. The ultimate receptor of these mitogenic signals is the retinoblastoma (Rb) family of pocket proteins, whose inactivation is a required step to license cell proliferation. However, little is known regarding the molecular events that connect Ras signaling with the cell cycle. Here, we provide genetic evidence to illustrate that the p53/p21 Cdk-interacting protein 1 (Cip1)/Rb axis is an essential component of the Ras signaling pathway. Indeed, knockdown of p53, p21Cip1, or Rb restores proliferative properties in cells arrested by ablation of the three Ras loci, H-, N- and K-Ras. Ras signaling selectively inactivates p53-mediated induction of p21Cip1 expression by inhibiting acetylation of specific lysine residues in the p53 DNA binding domain. Proliferation of cells lacking both Ras proteins and p53 can be prevented by reexpression of the human p53 ortholog, provided that it retains an active DNA binding domain and an intact lysine residue at position 164. These results unveil a previously unidentified role for p53 in preventing cell proliferation under unfavorable mitogenic conditions. Moreover, we provide evidence that cells lacking Ras and p53 proteins owe their proliferative properties to the unexpected retroactivation of the Raf/Mek/Erk cascade by a Ras-independent mechanism.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Malumbres M, Barbacid M. RAS oncogenes: The first 30 years. Nat Rev Cancer. 2003;3(6):459–465. - PubMed
-
- Drosten M, Lechuga CG, Barbacid M. Ras signaling is essential for skin development. Oncogene. 2014;33(22):2857–2865. - PubMed
-
- Scholl FA, et al. Mek1/2 MAPK kinases are essential for Mammalian development, homeostasis, and Raf-induced hyperplasia. Dev Cell. 2007;12(4):615–629. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
