

Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing
- PMID: 25288881
- PMCID: PMC4179624
- DOI: 10.4137/CIN.S13779
Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing
Abstract
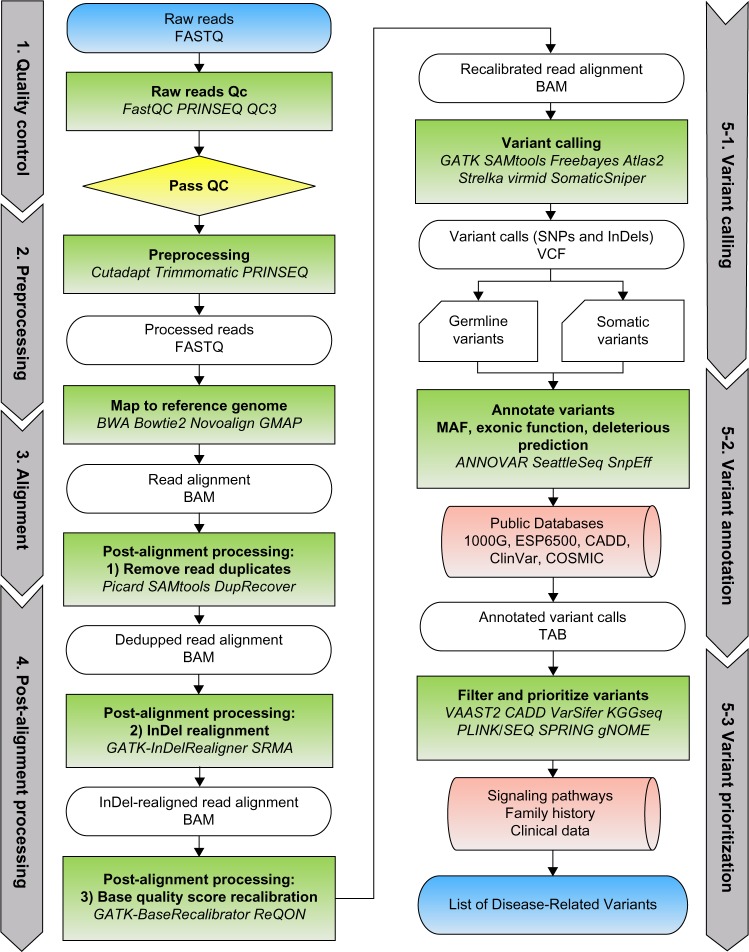
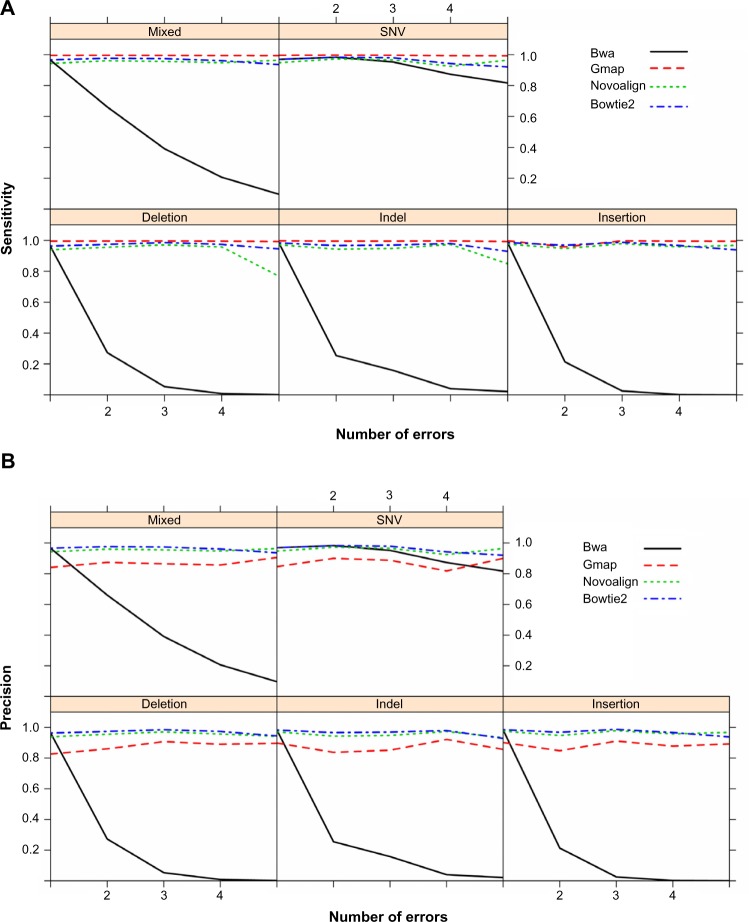
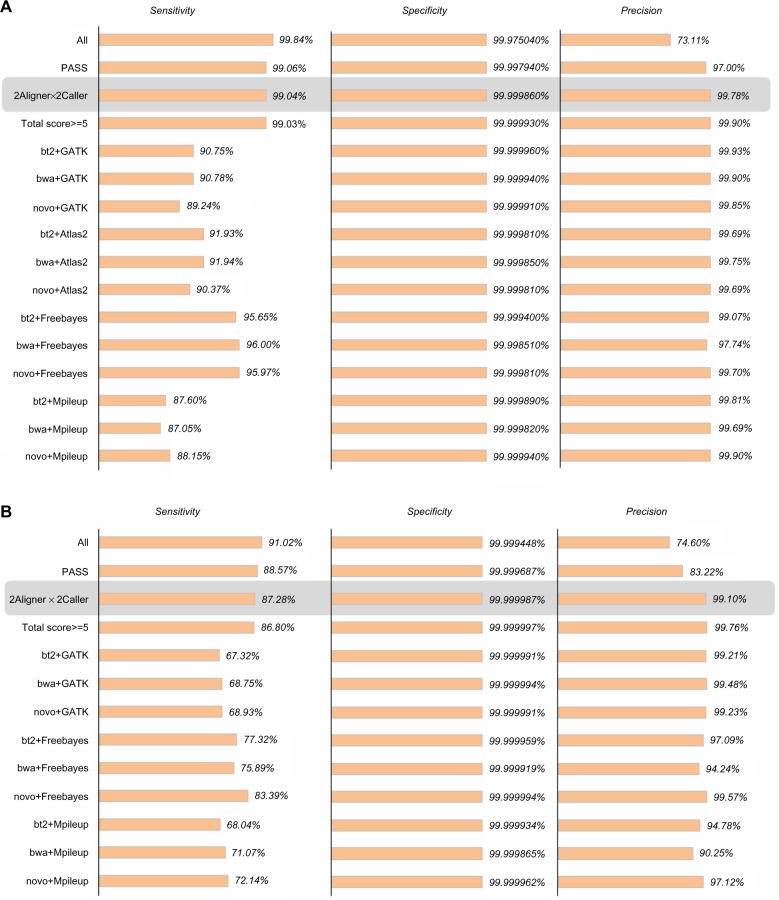
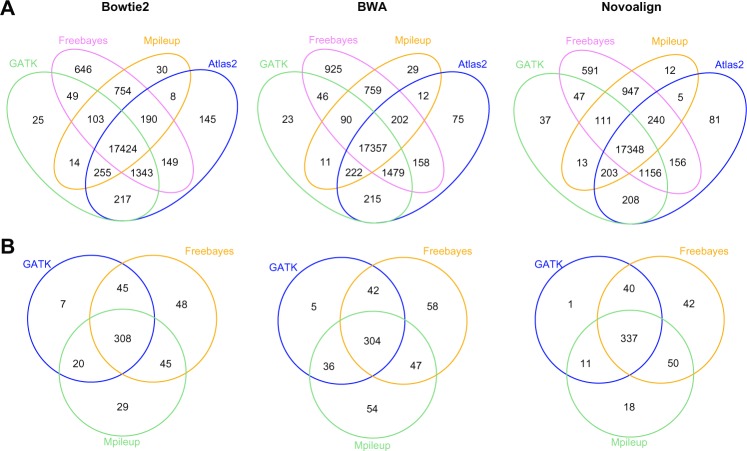
The advent of next-generation sequencing technologies has greatly promoted advances in the study of human diseases at the genomic, transcriptomic, and epigenetic levels. Exome sequencing, where the coding region of the genome is captured and sequenced at a deep level, has proven to be a cost-effective method to detect disease-causing variants and discover gene targets. In this review, we outline the general framework of whole exome sequence data analysis. We focus on established bioinformatics tools and applications that support five analytical steps: raw data quality assessment, pre-processing, alignment, post-processing, and variant analysis (detection, annotation, and prioritization). We evaluate the performance of open-source alignment programs and variant calling tools using simulated and benchmark datasets, and highlight the challenges posed by the lack of concordance among variant detection tools. Based on these results, we recommend adopting multiple tools and resources to reduce false positives and increase the sensitivity of variant calling. In addition, we briefly discuss the current status and solutions for big data management, analysis, and summarization in the field of bioinformatics.
Keywords: InDel; SNV; big data; next generation sequencing; sequence alignment; variant analysis; whole exome sequencing.
Figures
References
-
- Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–7. - PubMed
-
- Fertig EJ, Slebos R, Chung CH. Application of genomic and proteomic technologies in biomarker discovery. Am Soc Clin Oncol Educ Book. 2012;32:377–82. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
