

Cancer evolution: mathematical models and computational inference
- PMID: 25293804
- PMCID: PMC4265145
- DOI: 10.1093/sysbio/syu081
Cancer evolution: mathematical models and computational inference
Abstract
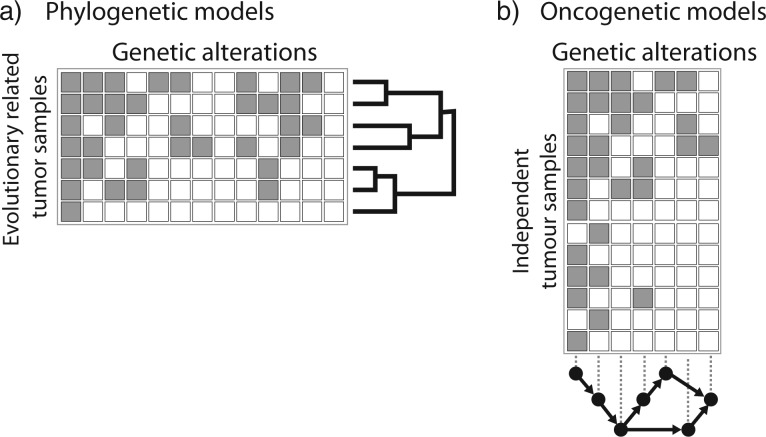
Cancer is a somatic evolutionary process characterized by the accumulation of mutations, which contribute to tumor growth, clinical progression, immune escape, and drug resistance development. Evolutionary theory can be used to analyze the dynamics of tumor cell populations and to make inference about the evolutionary history of a tumor from molecular data. We review recent approaches to modeling the evolution of cancer, including population dynamics models of tumor initiation and progression, phylogenetic methods to model the evolutionary relationship between tumor subclones, and probabilistic graphical models to describe dependencies among mutations. Evolutionary modeling helps to understand how tumors arise and will also play an increasingly important prognostic role in predicting disease progression and the outcome of medical interventions, such as targeted therapy.
Keywords: Cancer; cancer progression; evolution; population genetics; probabilistic graphical models.
© The Author(s) 2014. Published by Oxford University Press on behalf of the Society of Systematic Biologists.
Figures

References
-
- Adams R. P., Ghahramani Z., Jordan M. I. Tree-structured stick breaking processes for hierarchical data. Adv. Neural Inf. Process. Syst. (NIPS). 2010;23:19–27.
-
- Alarcón T., Byrne H., Maini P. A multiple scale model for tumor growth. Multiscale Model. Simul. 2005;3:440–475.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
