

Accounting for biases in riboprofiling data indicates a major role for proline in stalling translation
- PMID: 25294246
- PMCID: PMC4248317
- DOI: 10.1101/gr.175893.114
Accounting for biases in riboprofiling data indicates a major role for proline in stalling translation
Abstract
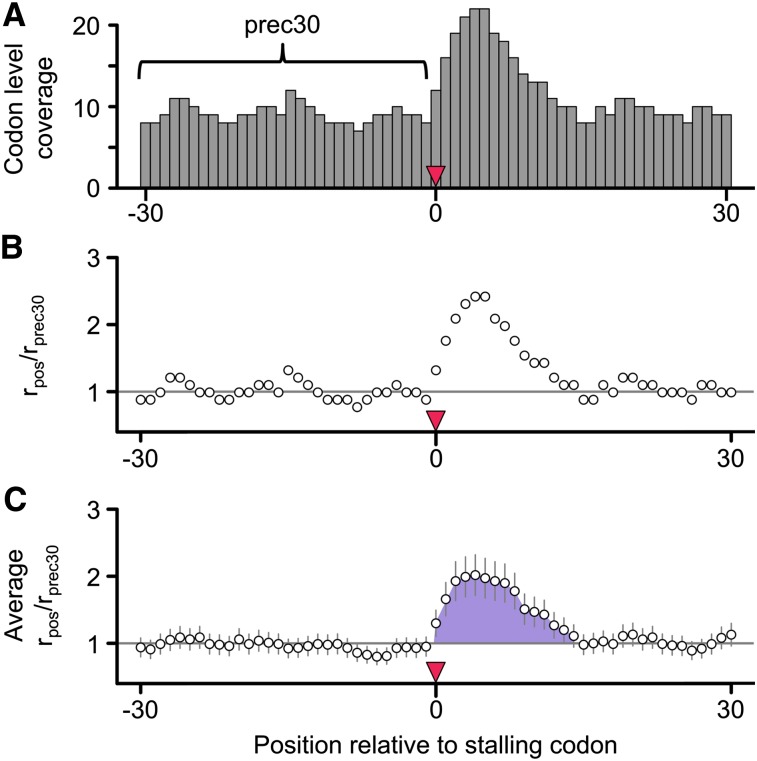
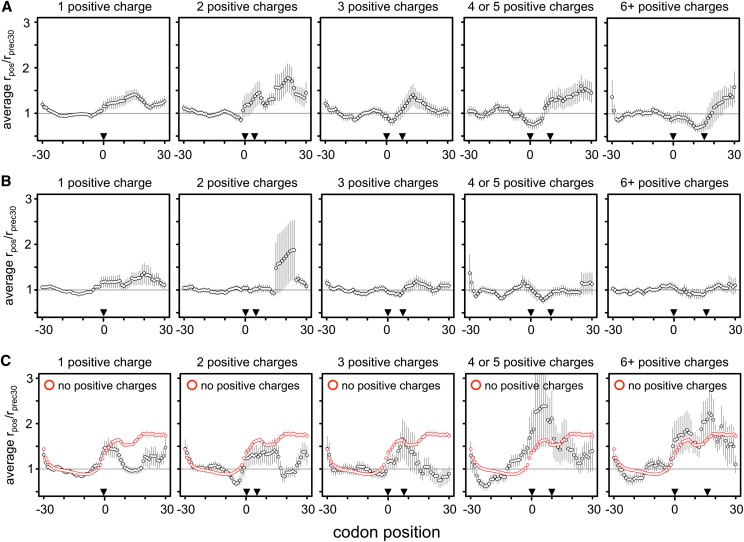
The recent advent of ribosome profiling-sequencing of short ribosome-bound fragments of mRNA-has offered an unprecedented opportunity to interrogate the sequence features responsible for modulating translational rates. Nevertheless, numerous analyses of the first riboprofiling data set have produced equivocal and often incompatible results. Here we analyze three independent yeast riboprofiling data sets, including two with much higher coverage than previously available, and find that all three show substantial technical sequence biases that confound interpretations of ribosomal occupancy. After accounting for these biases, we find no effect of previously implicated factors on ribosomal pausing. Rather, we find that incorporation of proline, whose unique side-chain stalls peptide synthesis in vitro, also slows the ribosome in vivo. We also reanalyze a method that implicated positively charged amino acids as the major determinant of ribosomal stalling and demonstrate that it produces false signals of stalling in low-coverage data. Our results suggest that any analysis of riboprofiling data should account for sequencing biases and sparse coverage. To this end, we establish a robust methodology that enables analysis of ribosome profiling data without prior assumptions regarding which positions spanned by the ribosome cause stalling.
© 2014 Artieri and Fraser; Published by Cold Spring Harbor Laboratory Press.
Figures
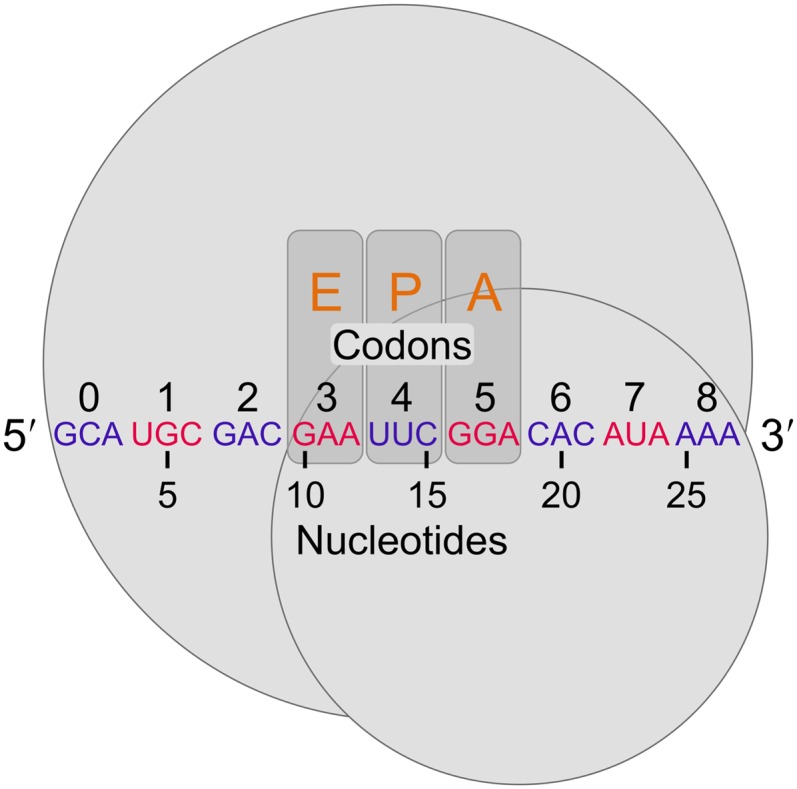
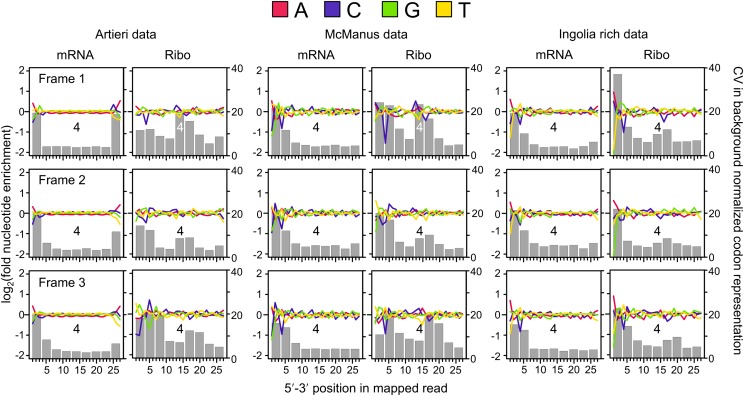
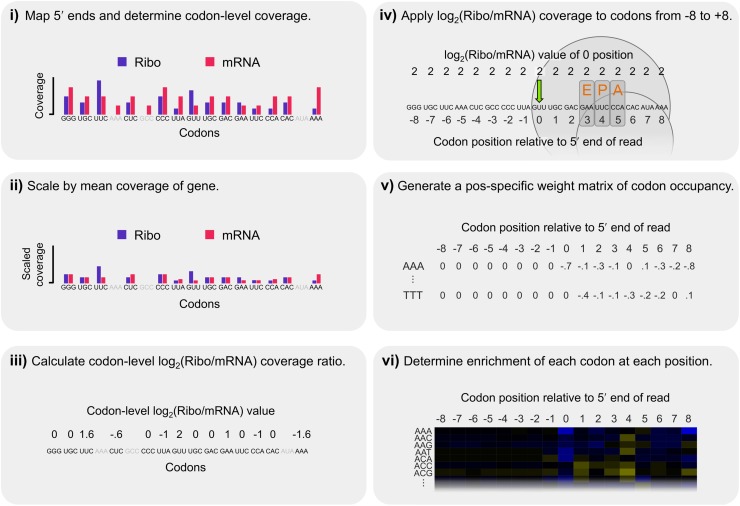
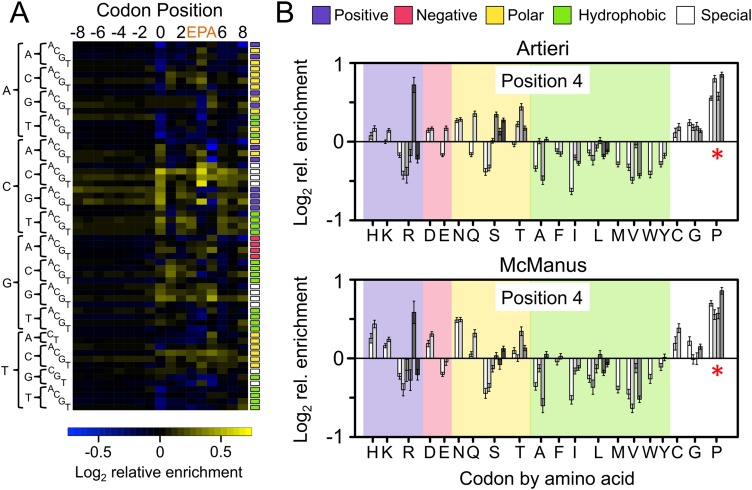
References
-
- Bennetzen JL, Hall BD. 1982. Codon selection in yeast. J Biol Chem 257: 3026–3031. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases