

Epigenetic signatures of alcohol abuse and hepatitis infection during human hepatocarcinogenesis
- PMID: 25294808
- PMCID: PMC4253444
- DOI: 10.18632/oncotarget.2444
Epigenetic signatures of alcohol abuse and hepatitis infection during human hepatocarcinogenesis
Abstract
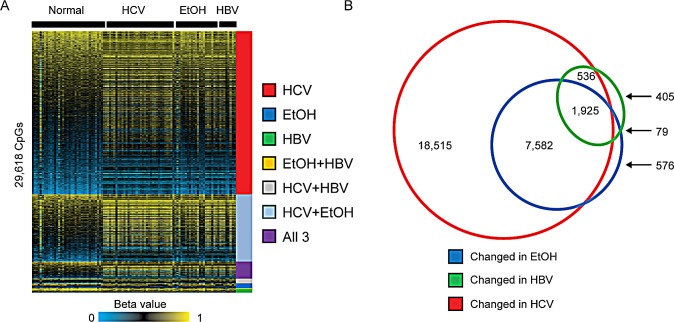
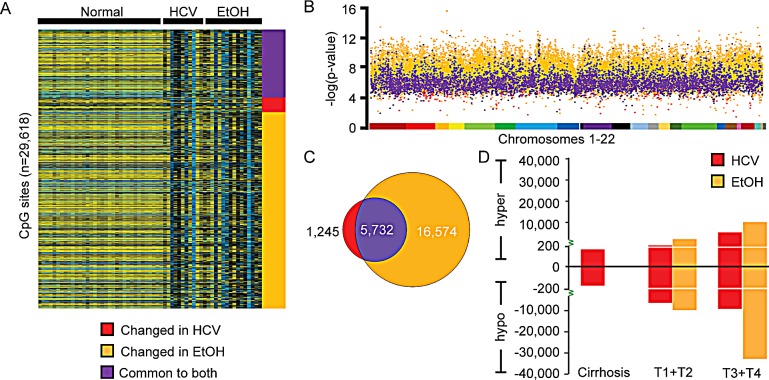
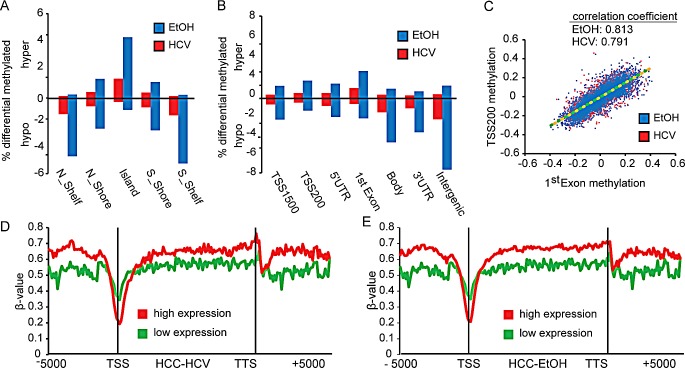
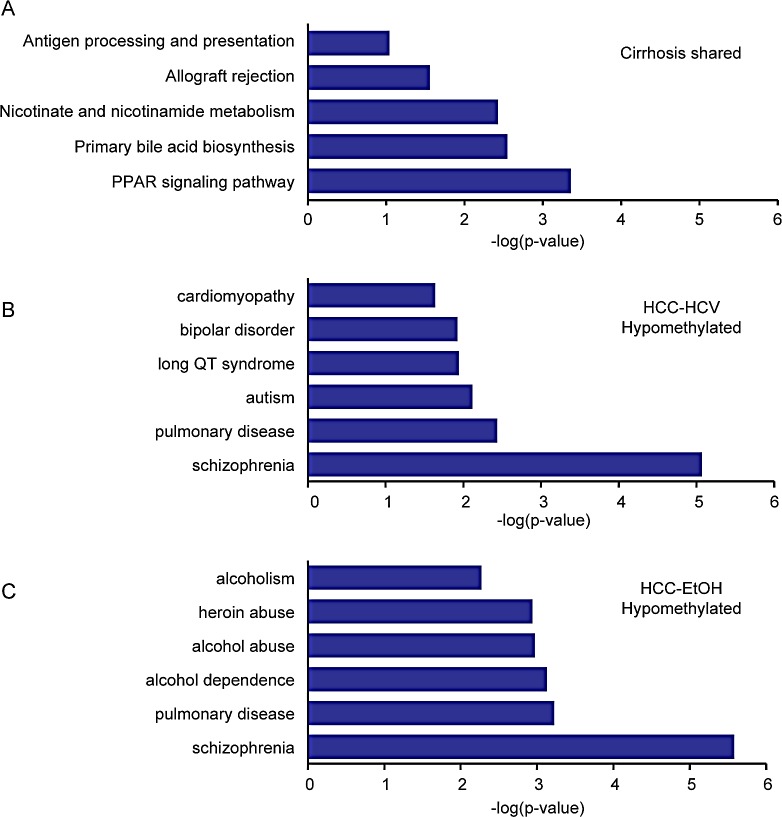
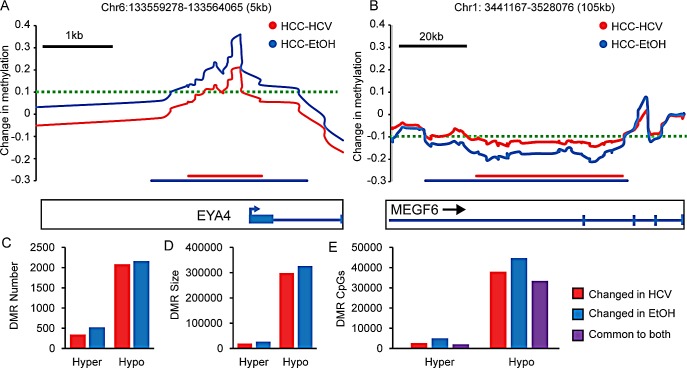
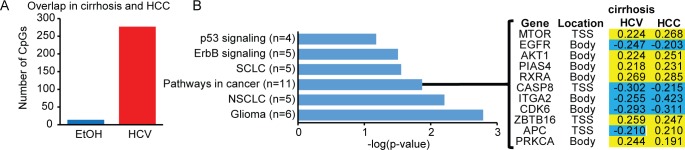
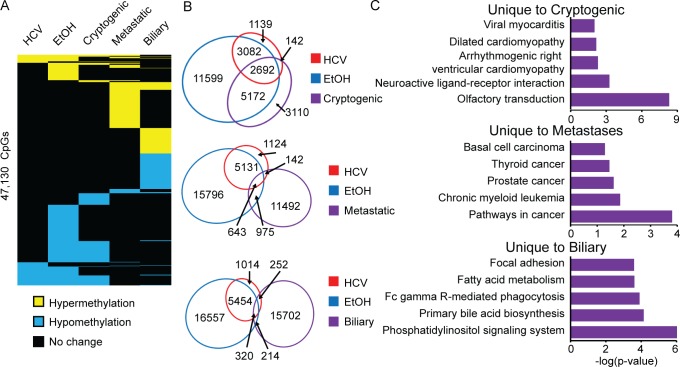
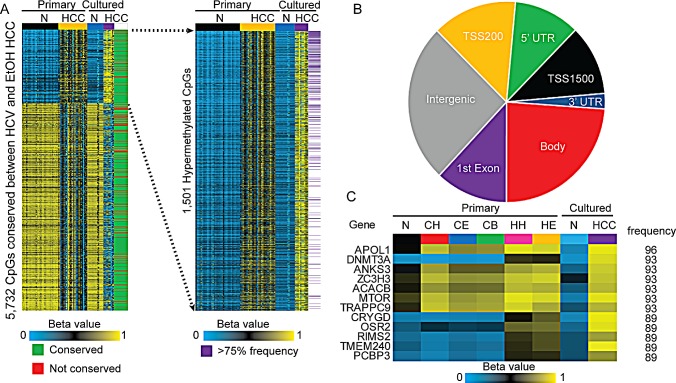
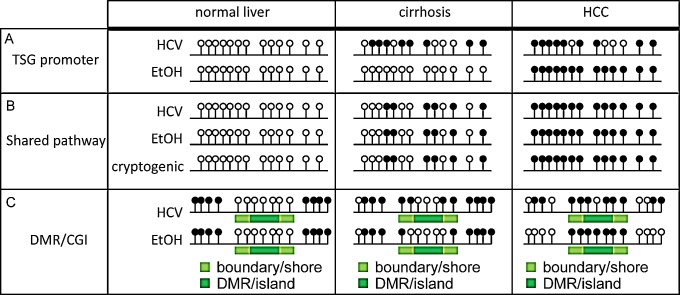
Hepatocellular carcinoma (HCC) is the second most common cause of cancer deaths worldwide. Deregulated DNA methylation landscapes are ubiquitous in human cancers. Interpretation of epigenetic aberrations in HCC is confounded by multiple etiologic drivers and underlying cirrhosis. We globally profiled the DNA methylome of 34 normal and 122 liver disease tissues arising in settings of hepatitis B (HBV) or C (HCV) viral infection, alcoholism (EtOH), and other causes to examine how these environmental agents impact DNA methylation in a manner that contributes to liver disease. Our results demonstrate that each 'exposure' leaves unique and overlapping signatures on the methylome. CpGs aberrantly methylated in cirrhosis-HCV and conserved in HCC were enriched for cancer driver genes, suggesting a pathogenic role for HCV-induced methylation changes. Additionally, large genomic regions displaying stepwise hypermethylation or hypomethylation during disease progression were identified. HCC-HCV/EtOH methylomes overlap highly with cryptogenic HCC, suggesting shared epigenetically deregulated pathways for hepatocarcinogenesis. Finally, overlapping methylation abnormalities between primary and cultured tumors unveil conserved epigenetic signatures in HCC. Taken together, this study reveals profound epigenome deregulation in HCC beginning during cirrhosis and influenced by common environmental agents. These results lay the foundation for defining epigenetic drivers and clinically useful methylation markers for HCC.
Figures
References
-
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. - PubMed
-
- Ferlay J SI, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet] Lyon, France: International Agency for Research on Cancer; 2013.
-
- El-Serag HB. Epidemiology of hepatocellular carcinoma in USA. Hepatol Res. 2007;37(Suppl 2):S88–94. - PubMed
-
- Bruix J, Boix L, Sala M, Llovet JM. Focus on hepatocellular carcinoma. Cancer Cell. 2004;5(3):215–219. - PubMed
-
- Marrero JA, Fontana RJ, Barrat A, Askari F, Conjeevaram HS, Su GL, Lok AS. Prognosis of hepatocellular carcinoma: comparison of 7 staging systems in an American cohort. Hepatology. 2005;41(4):707–716. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
