

svaseq: removing batch effects and other unwanted noise from sequencing data
- PMID: 25294822
- PMCID: PMC4245966
- DOI: 10.1093/nar/gku864
svaseq: removing batch effects and other unwanted noise from sequencing data
Abstract
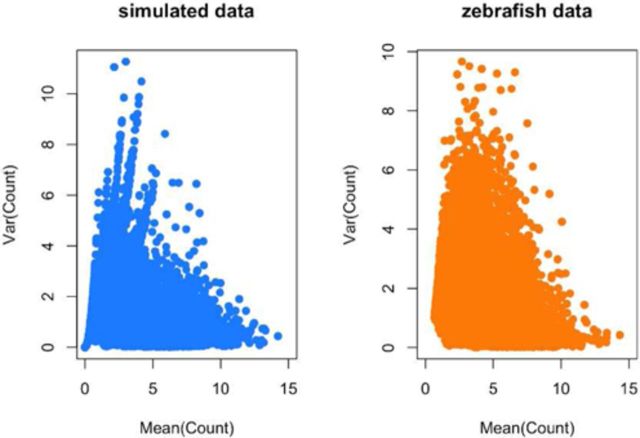
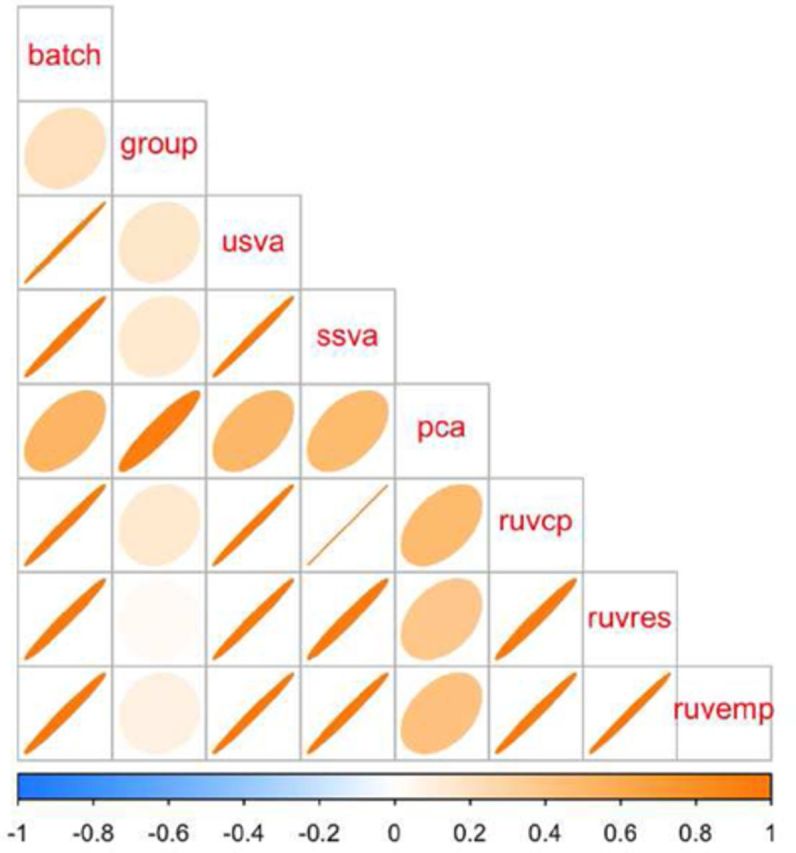
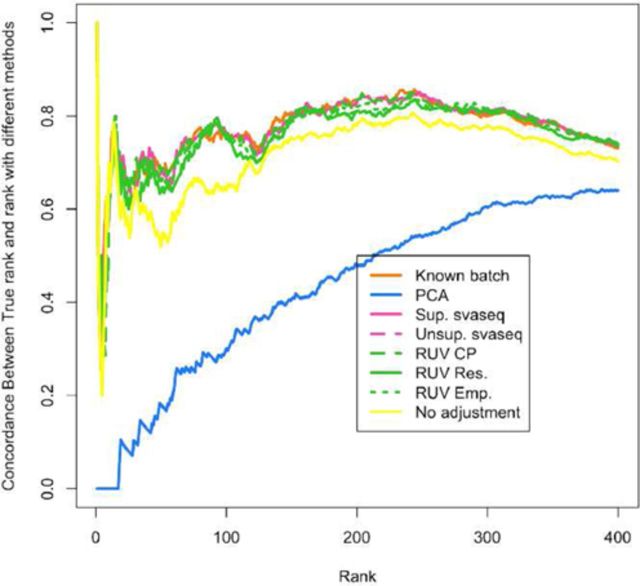
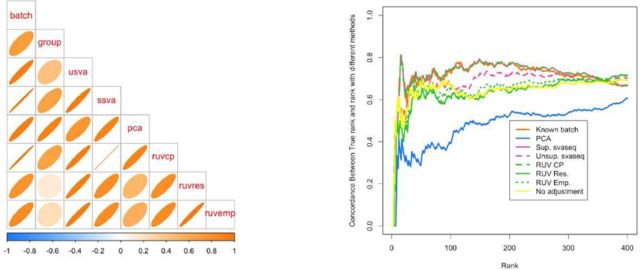
It is now known that unwanted noise and unmodeled artifacts such as batch effects can dramatically reduce the accuracy of statistical inference in genomic experiments. These sources of noise must be modeled and removed to accurately measure biological variability and to obtain correct statistical inference when performing high-throughput genomic analysis. We introduced surrogate variable analysis (sva) for estimating these artifacts by (i) identifying the part of the genomic data only affected by artifacts and (ii) estimating the artifacts with principal components or singular vectors of the subset of the data matrix. The resulting estimates of artifacts can be used in subsequent analyses as adjustment factors to correct analyses. Here I describe a version of the sva approach specifically created for count data or FPKMs from sequencing experiments based on appropriate data transformation. I also describe the addition of supervised sva (ssva) for using control probes to identify the part of the genomic data only affected by artifacts. I present a comparison between these versions of sva and other methods for batch effect estimation on simulated data, real count-based data and FPKM-based data. These updates are available through the sva Bioconductor package and I have made fully reproducible analysis using these methods available from: https://github.com/jtleek/svaseq.
© The Author(s) 2014. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures



References
-
- Akey J.M., Biswas S., Leek J.T., Storey J.D. On the design and analysis of gene expression studies in human populations. Nat. Genet. 2007;39:807–808. - PubMed
-
- Sebastiani P., Solovieff N., Puca A., Hartley S.W., Melista E., Andersen S., Dworkis D.A., Wilk J.B., Myers R.H., Steinberg M.H., et al. Genetic signatures of exceptional longevity in humans. Science. 2010;2010 doi:10.1126/science.1190532. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
