

Explicit DNase sequence bias modeling enables high-resolution transcription factor footprint detection
- PMID: 25294828
- PMCID: PMC4231734
- DOI: 10.1093/nar/gku810
Explicit DNase sequence bias modeling enables high-resolution transcription factor footprint detection
Abstract
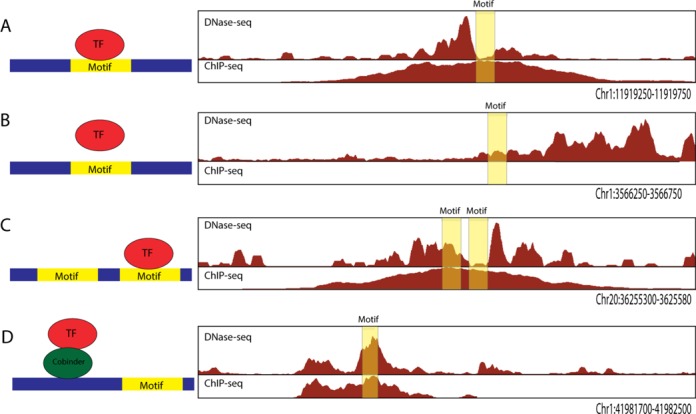
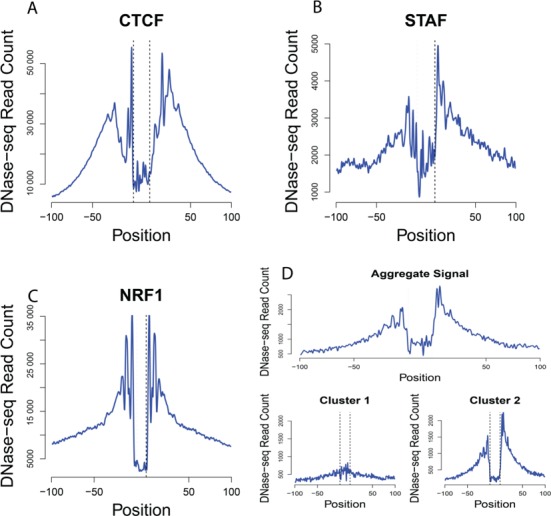
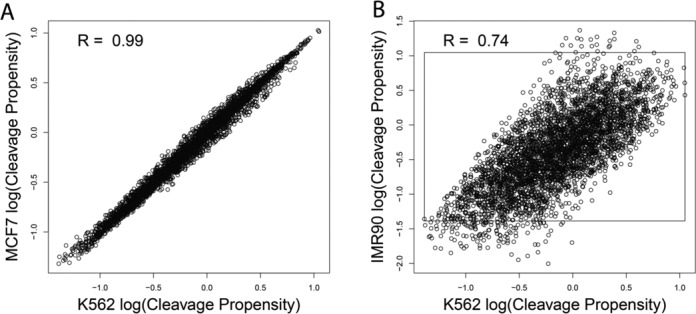
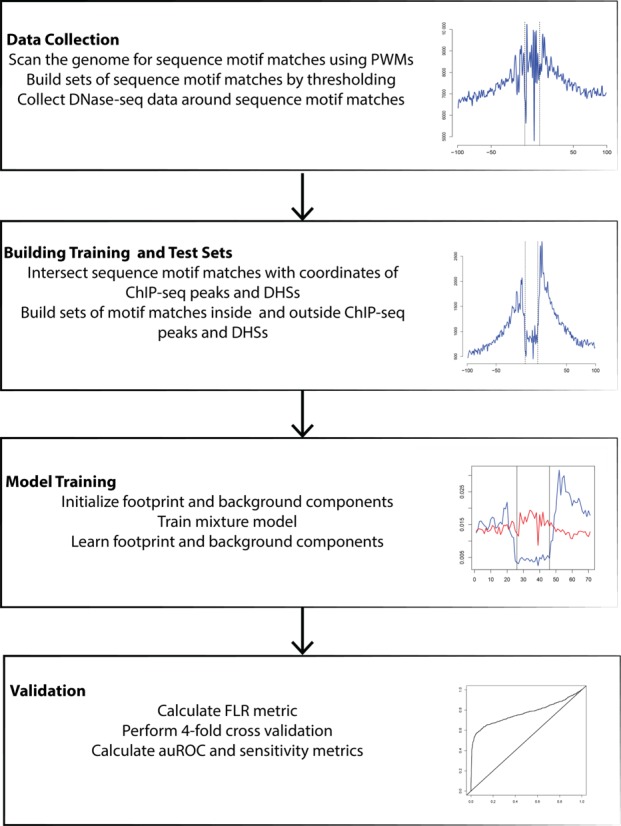
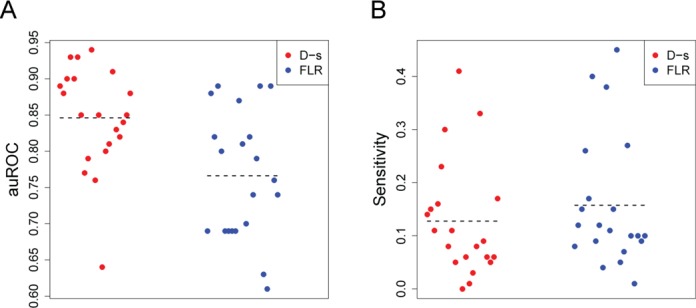
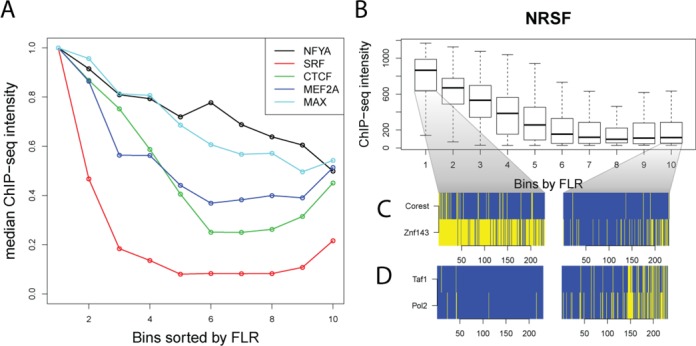
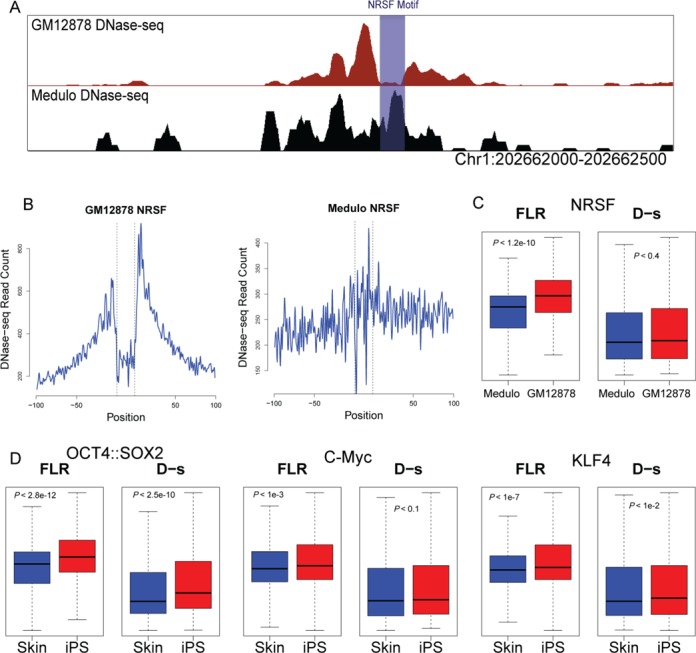
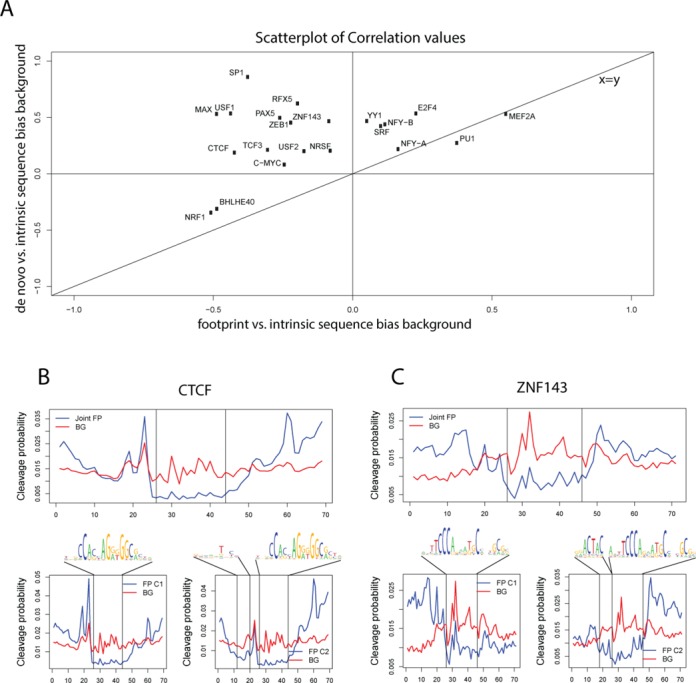
DNaseI footprinting is an established assay for identifying transcription factor (TF)-DNA interactions with single base pair resolution. High-throughput DNase-seq assays have recently been used to detect in vivo DNase footprints across the genome. Multiple computational approaches have been developed to identify DNase-seq footprints as predictors of TF binding. However, recent studies have pointed to a substantial cleavage bias of DNase and its negative impact on predictive performance of footprinting. To assess the potential for using DNase-seq to identify individual binding sites, we performed DNase-seq on deproteinized genomic DNA and determined sequence cleavage bias. This allowed us to build bias corrected and TF-specific footprint models. The predictive performance of these models demonstrated that predicted footprints corresponded to high-confidence TF-DNA interactions. DNase-seq footprints were absent under a fraction of ChIP-seq peaks, which we show to be indicative of weaker binding, indirect TF-DNA interactions or possible ChIP artifacts. The modeling approach was also able to detect variation in the consensus motifs that TFs bind to. Finally, cell type specific footprints were detected within DNase hypersensitive sites that are present in multiple cell types, further supporting that footprints can identify changes in TF binding that are not detectable using other strategies.
© The Author(s) 2014. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
