

c-Abl modulates tumor cell sensitivity to antibody-dependent cellular cytotoxicity
- PMID: 25300860
- PMCID: PMC4258447
- DOI: 10.1158/2326-6066.CIR-14-0083
c-Abl modulates tumor cell sensitivity to antibody-dependent cellular cytotoxicity
Abstract
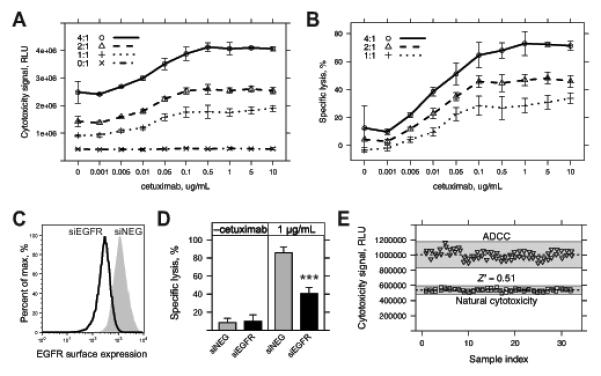
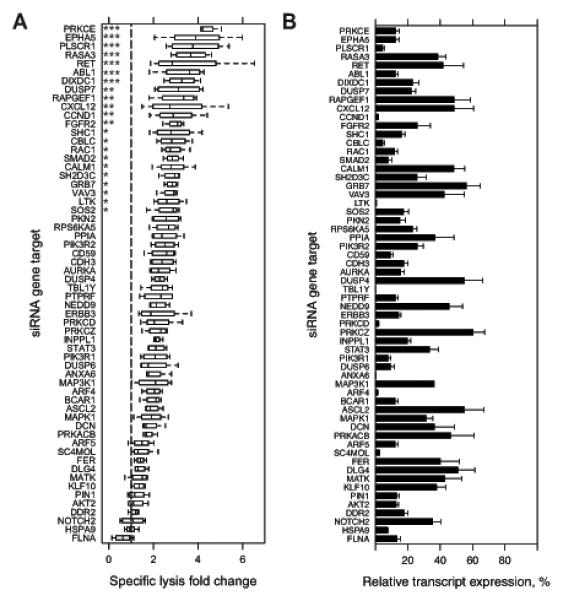
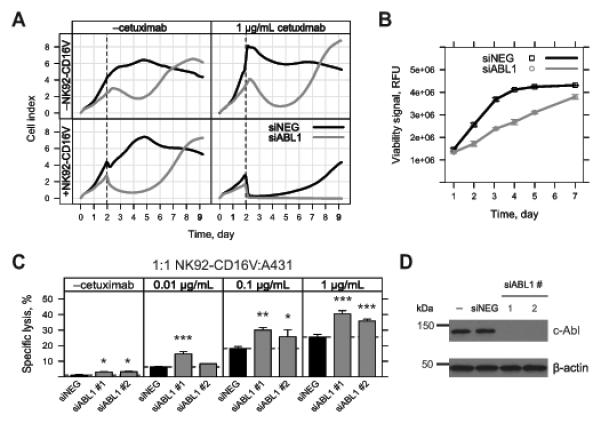
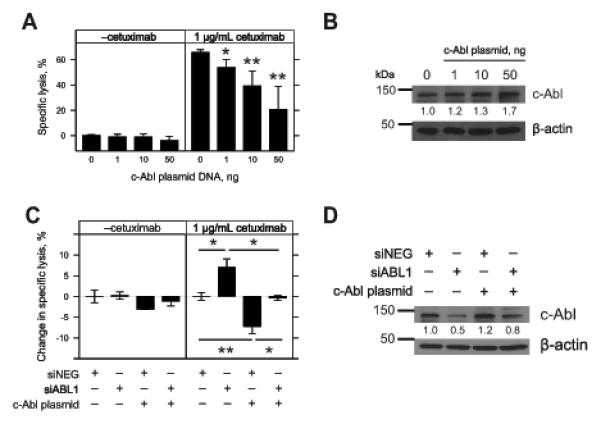
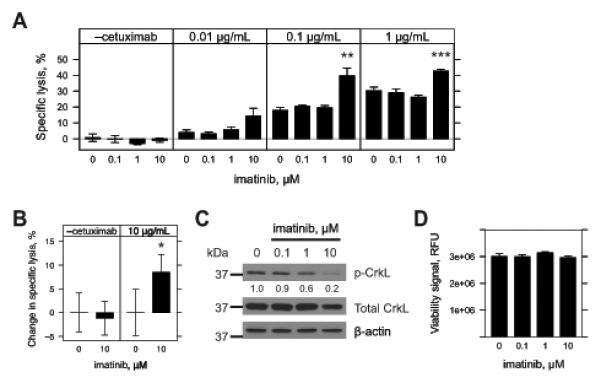
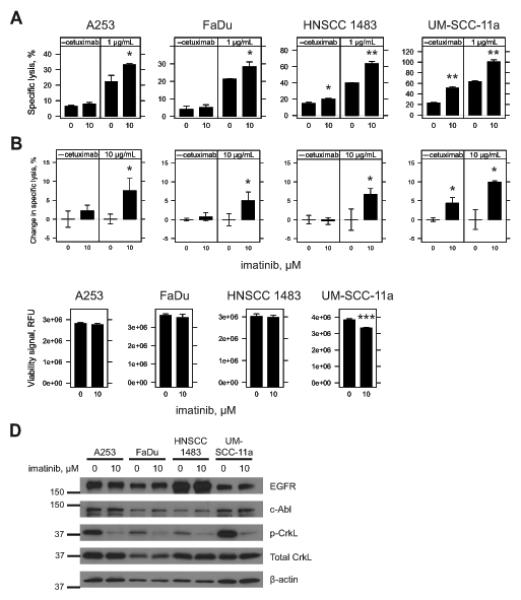
Monoclonal antibodies (mAb) can modulate cancer cell signal transduction and recruit antitumor immune effector mechanisms-including antibody-dependent cellular cytotoxicity (ADCC). Although several clinically effective antibodies can promote ADCC, therapeutic resistance is common. We hypothesized that oncogenic signaling networks within tumor cells affect their sensitivity to ADCC. We developed a screening platform and targeted 60 genes derived from an EGFR gene network using RNAi in an in vitro ADCC model system. Knockdown of GRB7, PRKCE, and ABL1 enhanced ADCC by primary and secondary screens. ABL1 knockdown also reduced cell proliferation, independent of its ADCC enhancement effects. c-Abl overexpression decreased ADCC sensitivity and rescued the effects of ABL1 knockdown. Imatinib inhibition of c-Abl kinase activity also enhanced ADCC-phenocopying ABL1 knockdown-against several EGFR-expressing head-and-neck squamous cell carcinoma cell lines by ex vivo primary natural killer cells. Our findings suggest that combining c-Abl inhibition with ADCC-promoting antibodies, such as cetuximab, could translate into increased therapeutic efficacy of mAbs.
©2014 American Association for Cancer Research.
Figures
References
-
- Wallace PK, Howell AL, Fanger MW. Role of Fc gamma receptors in cancer and infectious disease. J Leukoc Biol. 1994;55:816–26. - PubMed
-
- Chen Z, Freedman MS. CD16+ gammadelta T cells mediate antibody dependent cellular cytotoxicity: potential mechanism in the pathogenesis of multiple sclerosis. Clin Immunol. 2008;128:219–27. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
