

Current hypotheses on the mechanisms of alcoholism
- PMID: 25307591
- PMCID: PMC5898448
- DOI: 10.1016/B978-0-444-62619-6.00027-6
Current hypotheses on the mechanisms of alcoholism
Abstract
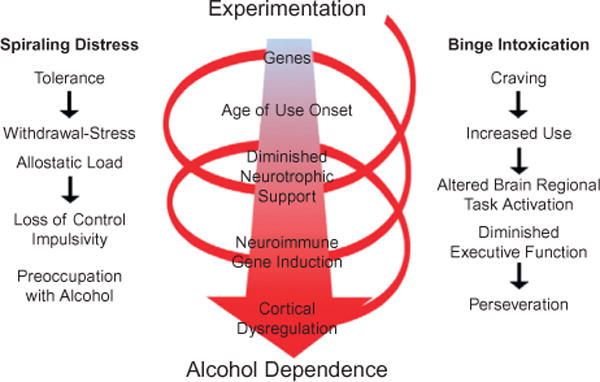
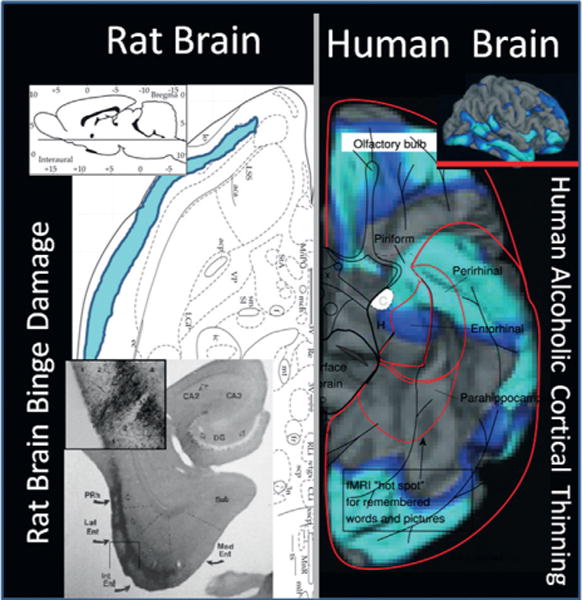
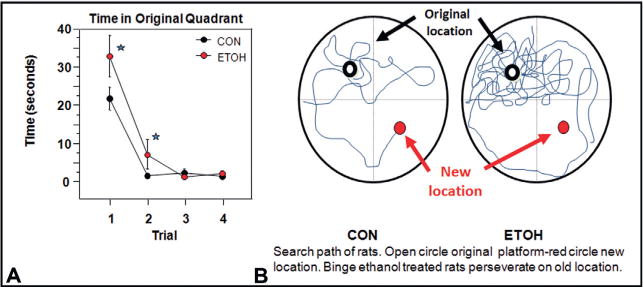
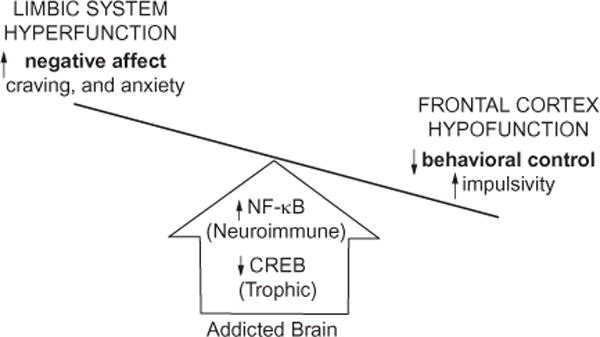
Chronic use of alcohol results in progressive changes to brain and behavior that often lead to the development of alcohol dependence and alcoholism. Although the mechanisms underlying the development of alcoholism remain to be fully elucidated, diminished executive functioning due to hypoactive prefrontal cortex executive control and hyperactive limbic system anxiety and negative emotion might contribute mechanistically to the shift from experimental use to alcoholism and dependence. In the chapter that follows, behavioral deficits associated with cortical dysfunction and neurodegeneration will be related to the behavioral characteristics of alcoholism (e.g., diminished executive function, impulsivity, altered limbic modulation). We will provide evidence that alterations in cyclic AMP-responsive element binding protein (CREB: neurotrophic) and NF-κB (neuroimmune) signaling contribute to the development and persistence of alcoholism. In addition, genetic predispositions and an earlier age of drinking onset will be discussed as contributing factors to the development of alcohol dependence and alcoholism. Overall chronic ethanol-induced neuroimmune gene induction is proposed to alter limbic and frontal neuronal networks contributing to the development and persistence of alcoholism.
Keywords: addiction; alcohol; anxiety; astrocyte; chemokines; cytokines; depression; human; innate immunity; microglia.
© 2014 Elsevier B.V. All rights reserved.
Figures
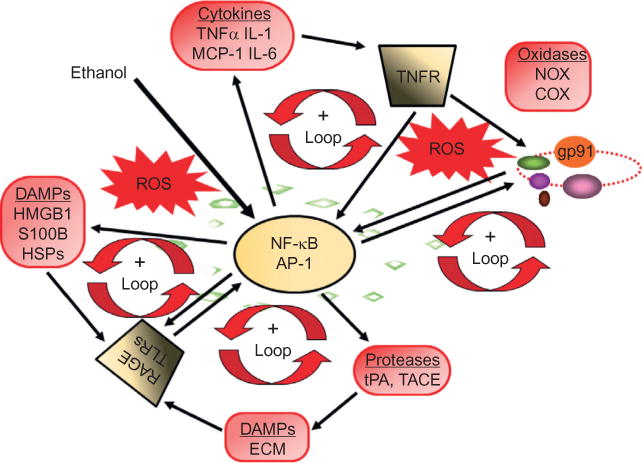
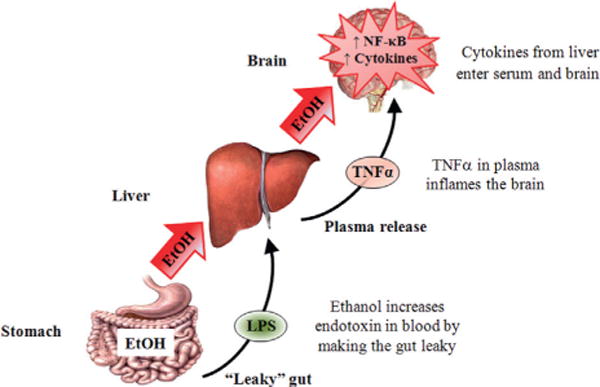
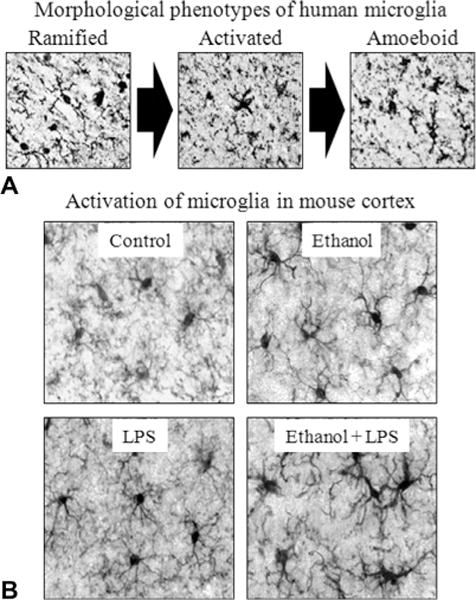
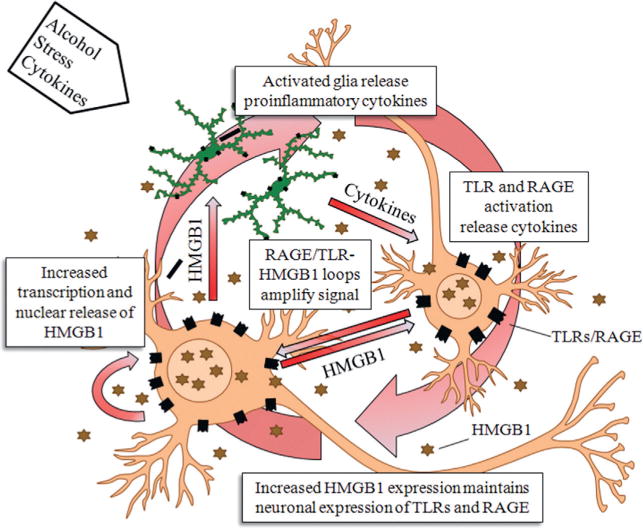
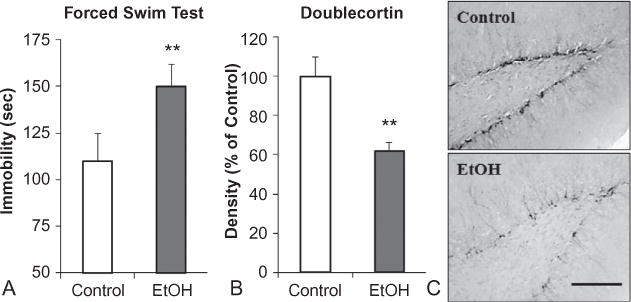
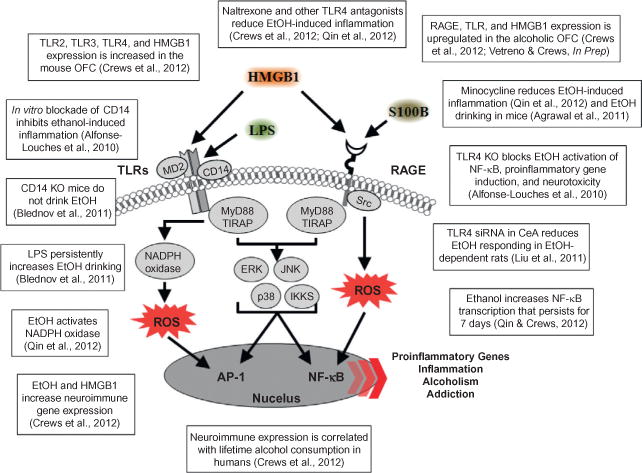
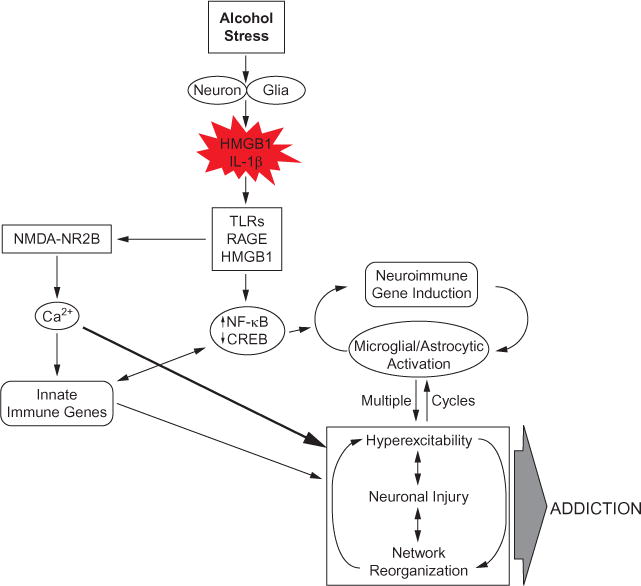
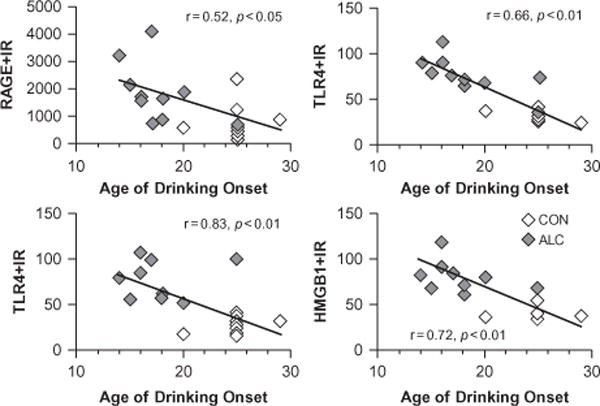
References
-
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. - PubMed
-
- Altman J, DAS GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965;124:319–335. - PubMed
-
- APA. Diagnostic and statistical manual of mental disorders: DSM-IV-TR. Washington, D.C: American Psychiatric Association; 2000.
-
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th. American Psychiatric Publishing; Arlington, VA: 2013.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
