

RNA structure analysis of alphacoronavirus terminal genome regions
- PMID: 25307890
- PMCID: PMC7114417
- DOI: 10.1016/j.virusres.2014.10.001
RNA structure analysis of alphacoronavirus terminal genome regions
Abstract
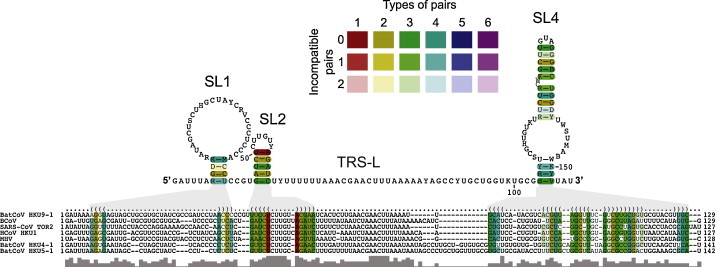
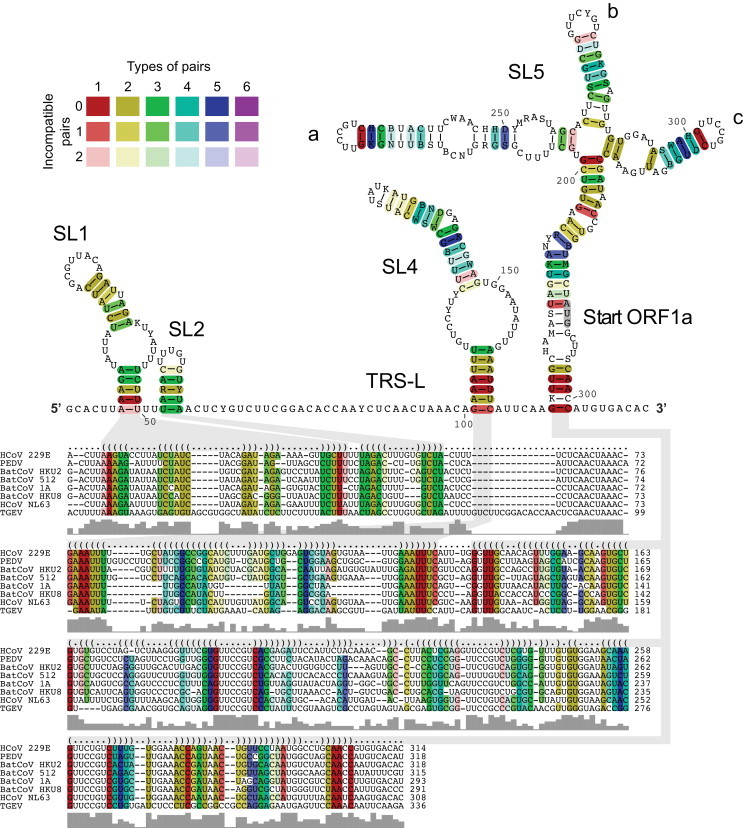
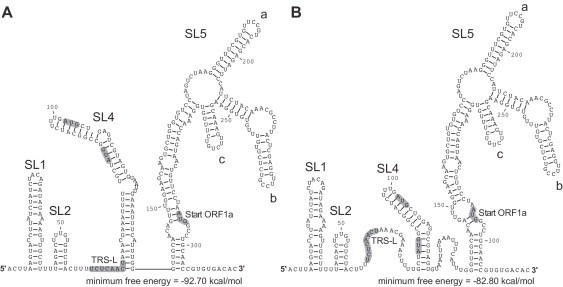
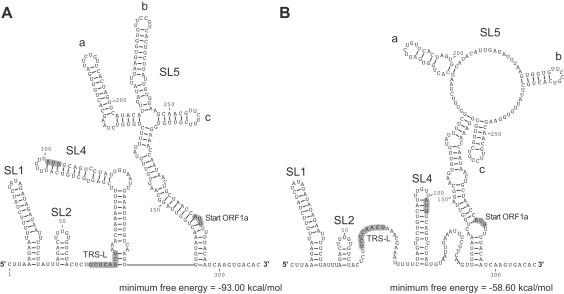
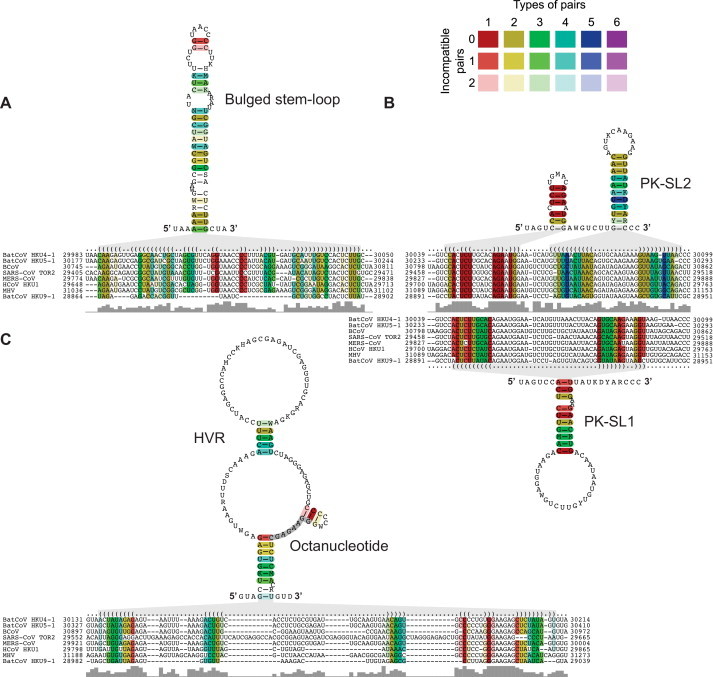
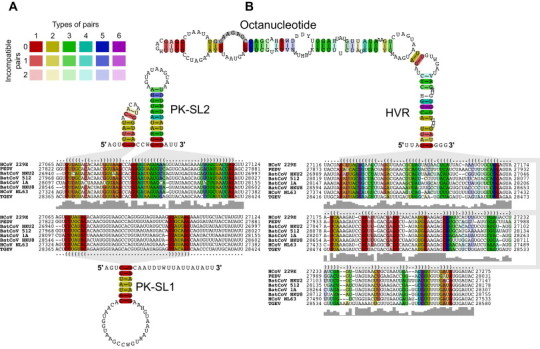
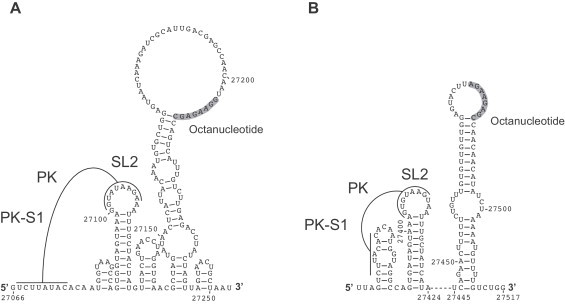
Coronavirus genome replication is mediated by a multi-subunit protein complex that is comprised of more than a dozen virally encoded and several cellular proteins. Interactions of the viral replicase complex with cis-acting RNA elements located in the 5' and 3'-terminal genome regions ensure the specific replication of viral RNA. Over the past years, boundaries and structures of cis-acting RNA elements required for coronavirus genome replication have been extensively characterized in betacoronaviruses and, to a lesser extent, other coronavirus genera. Here, we review our current understanding of coronavirus cis-acting elements located in the terminal genome regions and use a combination of bioinformatic and RNA structure probing studies to identify and characterize putative cis-acting RNA elements in alphacoronaviruses. The study suggests significant RNA structure conservation among members of the genus Alphacoronavirus but also across genus boundaries. Overall, the conservation pattern identified for 5' and 3'-terminal RNA structural elements in the genomes of alpha- and betacoronaviruses is in agreement with the widely used replicase polyprotein-based classification of the Coronavirinae, suggesting co-evolution of the coronavirus replication machinery with cognate cis-acting RNA elements.
Keywords: Coronavirus; RNA structure; RNA virus; Replication; cis-Acting element.
Copyright © 2014 Elsevier B.V. All rights reserved.
Figures
Similar articles
-
Structural and functional conservation of cis-acting RNA elements in coronavirus 5'-terminal genome regions.Virology. 2018 Apr;517:44-55. doi: 10.1016/j.virol.2017.11.025. Epub 2017 Dec 6. Virology. 2018. PMID: 29223446 Free PMC article.
-
Coronavirus cis-Acting RNA Elements.Adv Virus Res. 2016;96:127-163. doi: 10.1016/bs.aivir.2016.08.007. Epub 2016 Sep 6. Adv Virus Res. 2016. PMID: 27712622 Free PMC article. Review.
-
The structure and functions of coronavirus genomic 3' and 5' ends.Virus Res. 2015 Aug 3;206:120-33. doi: 10.1016/j.virusres.2015.02.025. Epub 2015 Feb 28. Virus Res. 2015. PMID: 25736566 Free PMC article. Review.
-
Defining the roles of cis-acting RNA elements in tombusvirus replicase assembly in vitro.J Virol. 2012 Jan;86(1):156-71. doi: 10.1128/JVI.00404-11. Epub 2011 Oct 19. J Virol. 2012. PMID: 22013057 Free PMC article.
-
Alpha- and betacoronavirus cis-acting RNA elements.Curr Opin Microbiol. 2024 Jun;79:102483. doi: 10.1016/j.mib.2024.102483. Epub 2024 May 8. Curr Opin Microbiol. 2024. PMID: 38723345 Review.
Cited by
-
Duplex formation between the template and the nascent strand in the transcription-regulating sequences is associated with the site of template switching in SARS - CoV-2.RNA Biol. 2021 Oct 15;18(sup1):148-156. doi: 10.1080/15476286.2021.1975388. Epub 2021 Sep 20. RNA Biol. 2021. PMID: 34541994 Free PMC article.
-
Structural and functional conservation of cis-acting RNA elements in coronavirus 5'-terminal genome regions.Virology. 2018 Apr;517:44-55. doi: 10.1016/j.virol.2017.11.025. Epub 2017 Dec 6. Virology. 2018. PMID: 29223446 Free PMC article.
-
Women in the European Virus Bioinformatics Center.Viruses. 2022 Jul 12;14(7):1522. doi: 10.3390/v14071522. Viruses. 2022. PMID: 35891501 Free PMC article.
-
In silico identification of conserved cis-acting RNA elements in the SARS-CoV-2 genome.Future Virol. 2020 Jul;15(7):409-417. doi: 10.2217/fvl-2020-0163. Future Virol. 2020. PMID: 33005212 Free PMC article.
-
Exploiting Translation Machinery for Cancer Therapy: Translation Factors as Promising Targets.Int J Mol Sci. 2024 Oct 9;25(19):10835. doi: 10.3390/ijms251910835. Int J Mol Sci. 2024. PMID: 39409166 Free PMC article. Review.
References
-
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
