

A comparison of control samples for ChIP-seq of histone modifications
- PMID: 25309581
- PMCID: PMC4174756
- DOI: 10.3389/fgene.2014.00329
A comparison of control samples for ChIP-seq of histone modifications
Abstract
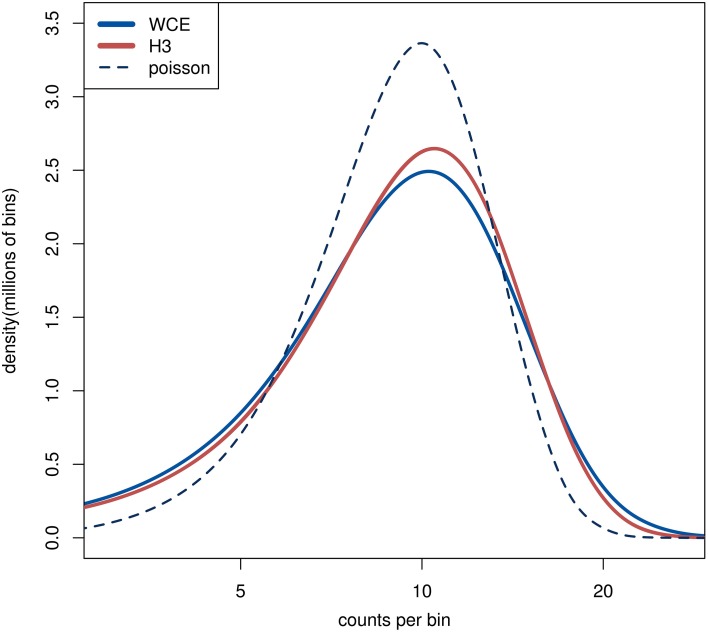
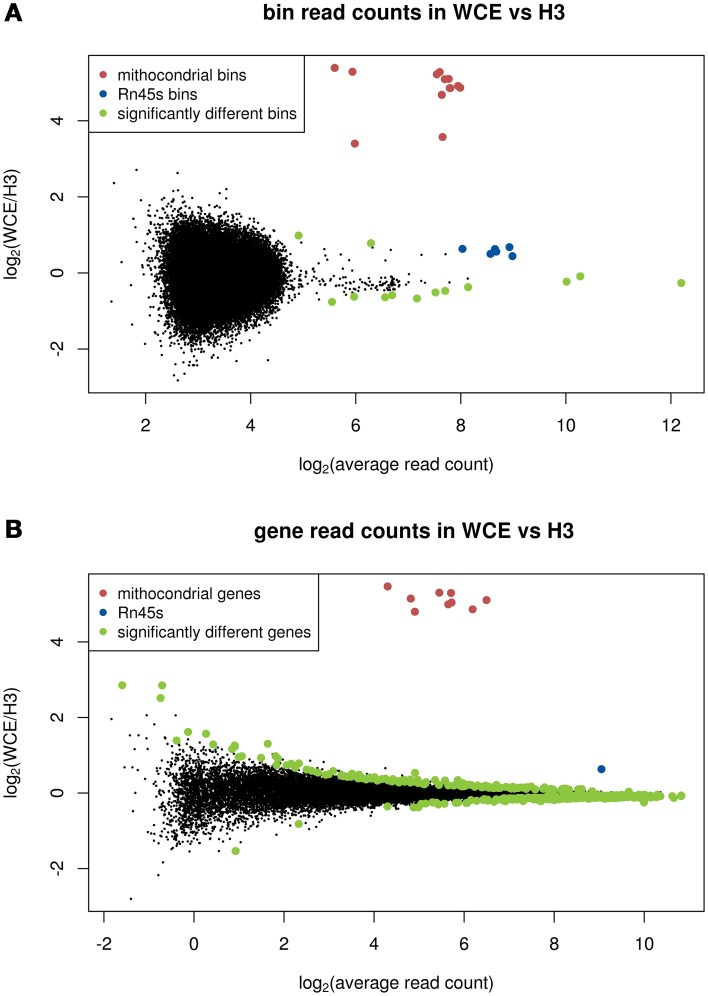
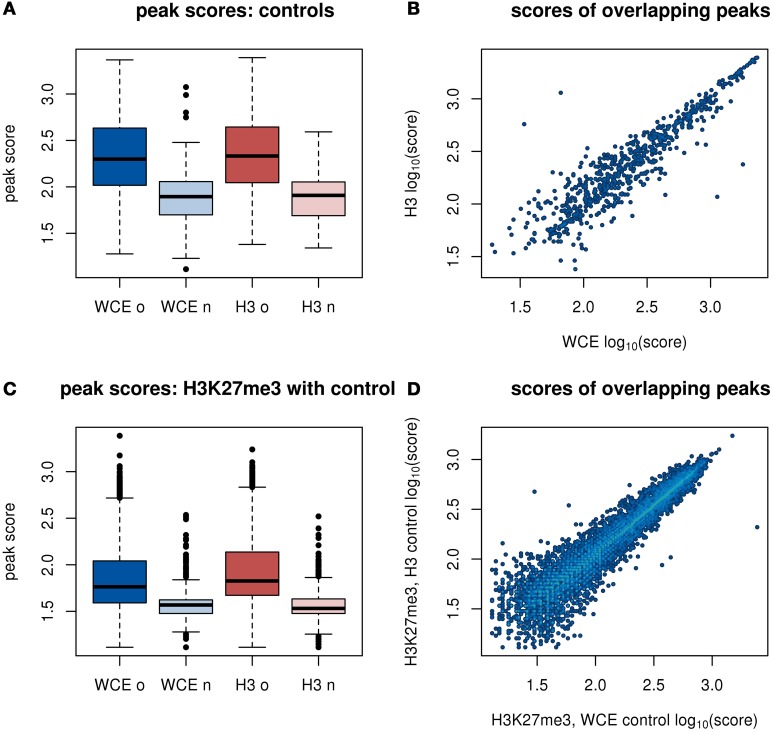
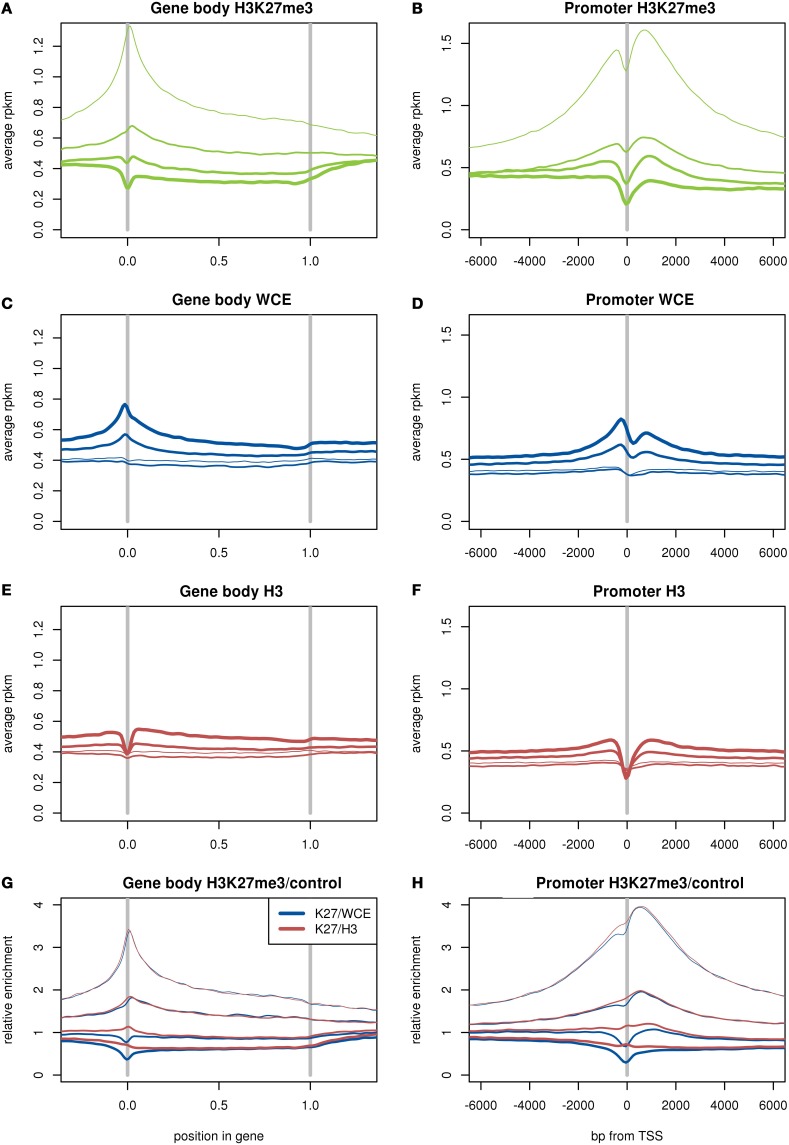
The advent of high-throughput sequencing has allowed genome wide profiling of histone modifications by Chromatin ImmunoPrecipitation (ChIP) followed by sequencing (ChIP-seq). In this assay the histone mark of interest is enriched through a chromatin pull-down assay using an antibody for the mark. Due to imperfect antibodies and other factors, many of the sequenced fragments do not originate from the histone mark of interest, and are referred to as background reads. Background reads are not uniformly distributed and therefore control samples are usually used to estimate the background distribution at any given genomic position. The Encyclopedia of DNA Elements (ENCODE) Consortium guidelines suggest sequencing a whole cell extract (WCE, or "input") sample, or a mock ChIP reaction such as an IgG control, as a background sample. However, for a histone modification ChIP-seq investigation it is also possible to use a Histone H3 (H3) pull-down to map the underlying distribution of histones. In this paper we generated data from a hematopoietic stem and progenitor cell population isolated from mouse fetal liver to compare WCE and H3 ChIP-seq as control samples. The quality of the control samples is estimated by a comparison to pull-downs of histone modifications and to expression data. We find minor differences between WCE and H3 ChIP-seq, such as coverage in mitochondria and behavior close to transcription start sites. Where the two controls differ, the H3 pull-down is generally more similar to the ChIP-seq of histone modifications. However, the differences between H3 and WCE have a negligible impact on the quality of a standard analysis.
Keywords: ChIP-seq; H3; control sample; histone modifications; input; quality control; whole cell extract.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources