

Identification of clinical target areas in the brainstem of prion-infected mice
- PMID: 25311251
- PMCID: PMC4949711
- DOI: 10.1111/nan.12189
Identification of clinical target areas in the brainstem of prion-infected mice
Abstract
Aims: While prion infection ultimately involves the entire brain, it has long been thought that the abrupt clinical onset and rapid neurological decline in laboratory rodents relates to involvement of specific critical neuroanatomical target areas. The severity and type of clinical signs, together with the rapid progression, suggest the brainstem as a candidate location for such critical areas. In this study we aimed to correlate prion pathology with clinical phenotype in order to identify clinical target areas.
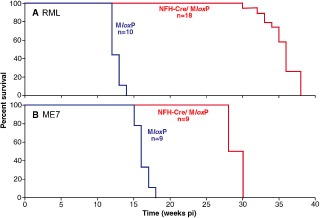
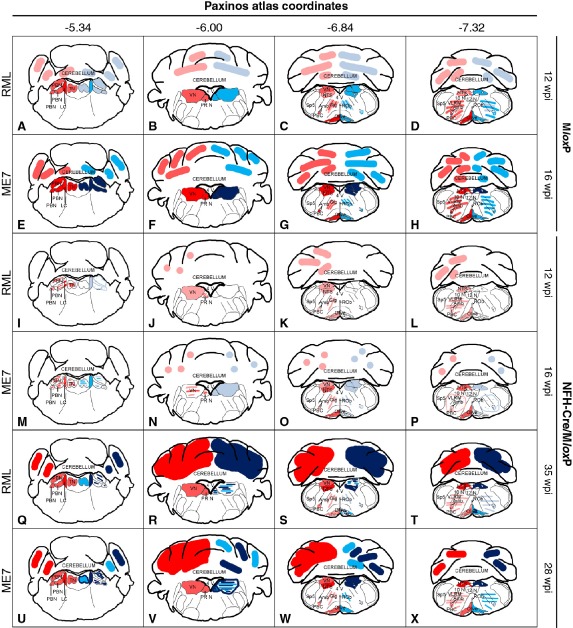
Method: We conducted a comprehensive survey of brainstem pathology in mice infected with two distinct prion strains, which produce different patterns of pathology, in mice overexpressing prion protein (with accelerated clinical onset) and in mice in which neuronal expression was reduced by gene targeting (which greatly delays clinical onset).
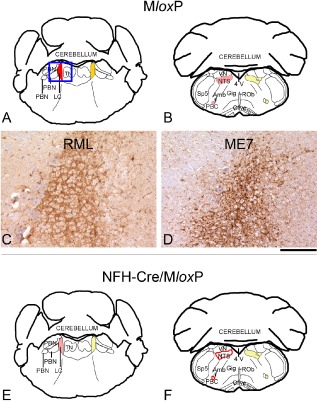
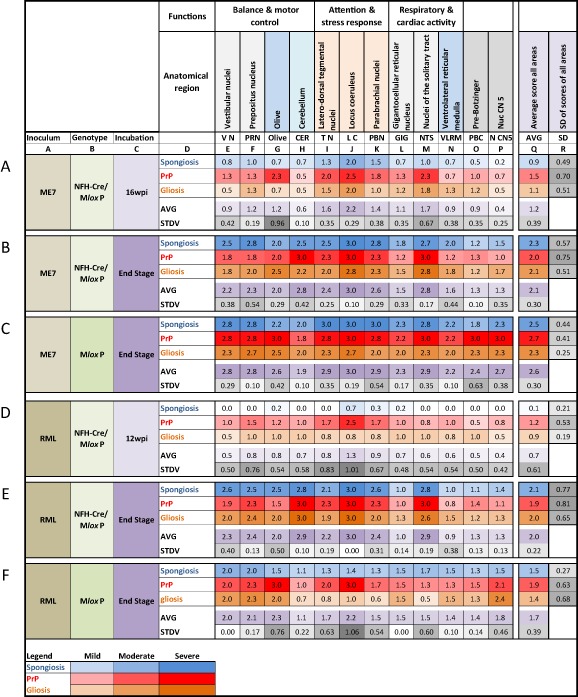
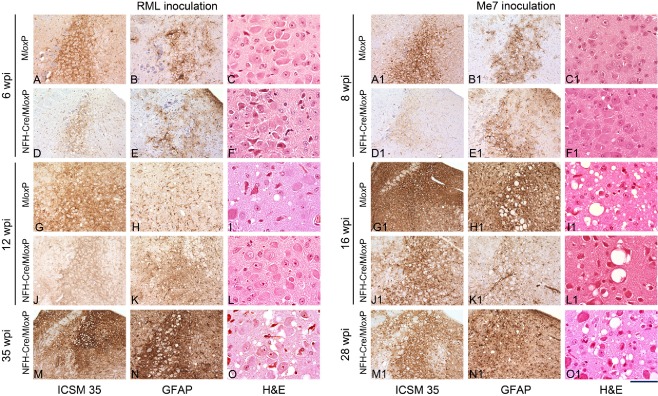
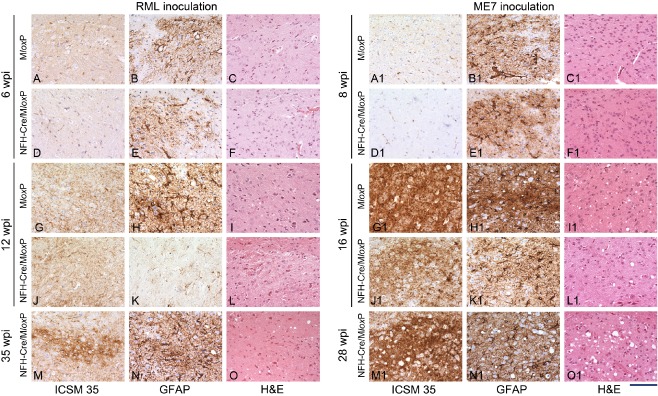
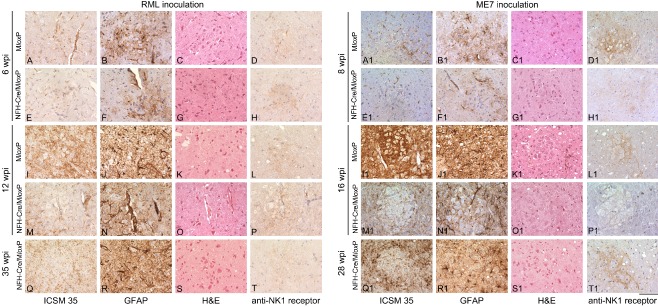
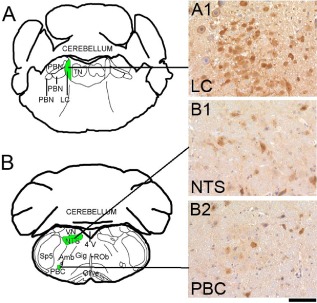
Results: We identified specific brainstem areas that are affected by prion pathology during the progression of the disease. In the early phase of disease the locus coeruleus, the nucleus of the solitary tract, and the pre-Bötzinger complex were affected by prion protein deposition. This was followed by involvement of the motor and autonomic centres of the brainstem.
Conclusions: Neurodegeneration in the locus coeruleus, the nucleus of the solitary tract and the pre-Bötzinger complex predominated and corresponded to the manifestation of the clinical phenotype. Because of their fundamental role in controlling autonomic function and the overlap with clinical signs in sporadic Creutzfeldt-Jakob disease, we suggest that these nuclei represent key clinical target areas in prion diseases.
Keywords: brainstem; clinical target areas; cre-lox system; locus coeruleus; neurodegeneration; prions.
© 2014 The Authors. Neuropathology and Applied Neurobiology published by John Wiley & Sons Ltd on behalf of British Neuropathological Society.
Figures
References
-
- Bruce ME, McBride PA, Farquhar CF. Precise targeting of the pathology of the sialoglycoprotein, PrP, and vacuolar degeneration in mouse scrapie. Neurosci Lett 1989; 102: 1–6 - PubMed
-
- Bruce ME. Serial studies on the development of cerebral amyloidosis and vacuolar degeneration in murine scrapie. J Comp Pathol 1981; 91: 589–597 - PubMed
-
- Liberski PP, Sikorska B, Hauw JJ, Kopp N, Streichenberger N, Giraud P, Boellaard J, Budka H, Kovacs GG, Ironside J, Brown P. Ultrastructural characteristics (or evaluation) of Creutzfeldt‐Jakob disease and other human transmissible spongiform encephalopathies or prion diseases. Ultrastruct Pathol 2010; 34: 351–361 - PubMed
-
- Liberski PP, Ironside JW. An outline of the neuropathology of transmissible spongiform encephalopathies (prion diseases). Folia Neuropathol 2004; 42 (Suppl. B): 39–58 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
