

Beyond the whole genome consensus: unravelling of PRRSV phylogenomics using next generation sequencing technologies
- PMID: 25312450
- PMCID: PMC4275598
- DOI: 10.1016/j.virusres.2014.10.004
Beyond the whole genome consensus: unravelling of PRRSV phylogenomics using next generation sequencing technologies
Abstract
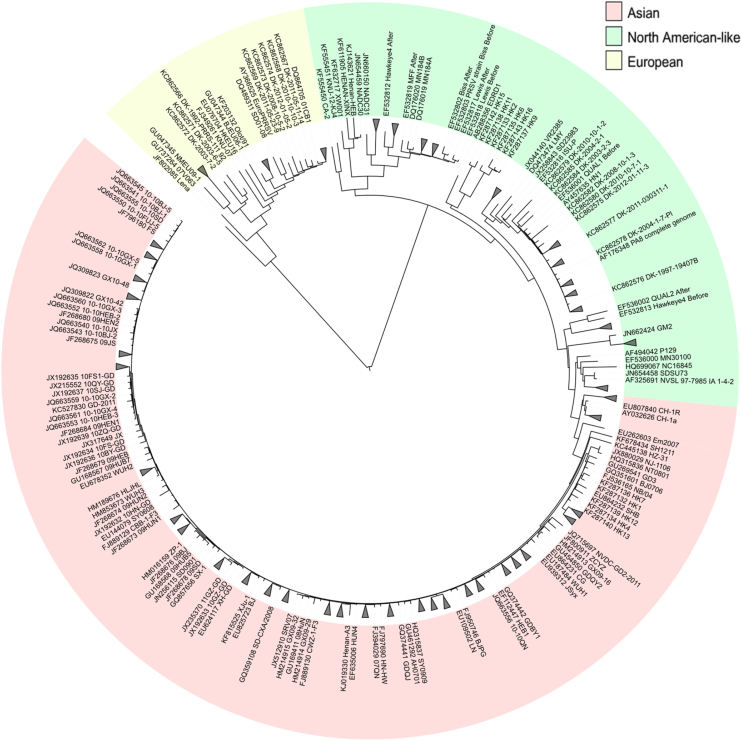
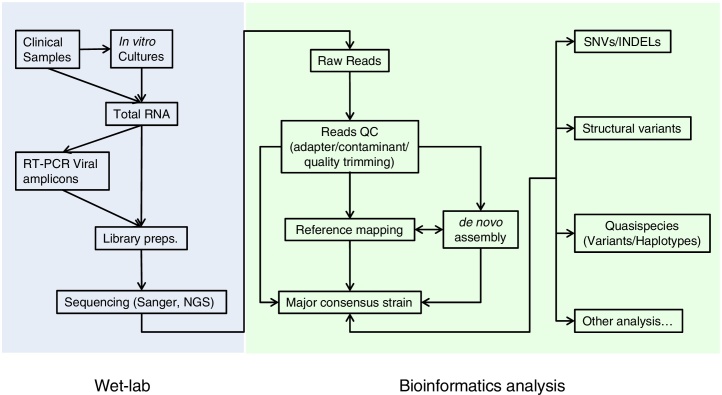
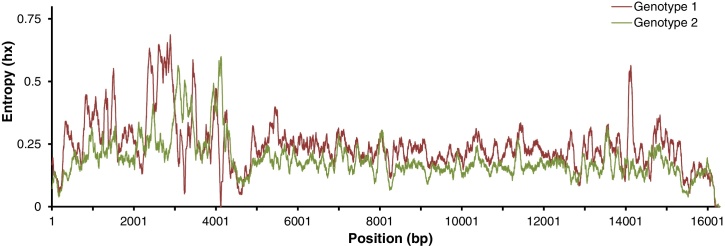
The highly heterogeneous porcine reproductive and respiratory syndrome virus (PRRSV) is the causative agent responsible for an economically important pig disease with the characteristic symptoms of reproductive losses in breeding sows and respiratory illnesses in young piglets. The virus can be broadly divided into the European and North American-like genotype 1 and 2 respectively. In addition to this intra-strains variability, the impact of coexisting viral quasispecies on disease development has recently gained much attention; owing very much to the advent of the next-generation sequencing (NGS) technologies. Genomic data produced from the massive sequencing capacities of NGS have enabled the study of PRRSV at an unprecedented rate and details. Unlike conventional sequencing methods which require knowledge of conserved regions, NGS allows de novo assembly of the full viral genomes. Evolutionary variations gained from different genotypic strains provide valuable insights into functionally important regions of the virus. Together with the advancement of sophisticated bioinformatics tools, ultra-deep NGS technologies make the detection of low frequency co-evolving quasispecies possible. This short review gives an overview, including a proposed workflow, on the use of NGS to explore the genetic diversity of PRRSV at both macro- and micro-evolutionary levels.
Keywords: PRRSV; Phylogenomics; Quasispecies; Ultra-deep next generation sequencing.
Copyright © 2014 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Allende R., Lewis T.L., Lu Z., Rock D.L., Kutish G.F., Ali A., Doster A.R., Osorio F.A. North American and European porcine reproductive and respiratory syndrome viruses differ in non-structural protein coding regions. J. Gen. Virol. 1999;80(Pt 2):307–315. - PubMed
-
- Andrews S. 2010. FastQC: A Quality Control Tool for High Throughput Sequence Data.http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
- Barzon L., Lavezzo E., Costanzi G., Franchin E., Toppo S., Palu G. Next-generation sequencing technologies in diagnostic virology. J. Clin. Virol. 2013;58(2):346–350. - PubMed
-
- Bashir A., Klammer A.A., Robins W.P., Chin C.S., Webster D., Paxinos E., Hsu D., Ashby M., Wang S., Peluso P., Sebra R., Sorenson J., Bullard J., Yen J., Valdovino M., Mollova E., Luong K., Lin S., LaMay B., Joshi A., Rowe L., Frace M., Tarr C.L., Turnsek M., Davis B.M., Kasarskis A., Mekalanos J.J., Waldor M.K., Schadt E.E. A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotechnol. 2012;30(7):701–707. - PMC - PubMed
-
- Beerenwinkel N., Zagordi O. Ultra-deep sequencing for the analysis of viral populations. Curr. Opin. Virol. 2011;1(5):413–418. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
