

Phylostratigraphic bias creates spurious patterns of genome evolution
- PMID: 25312911
- PMCID: PMC4271527
- DOI: 10.1093/molbev/msu286
Phylostratigraphic bias creates spurious patterns of genome evolution
Erratum in
-
Phylostratigraphic Bias Creates Spurious Patterns of Genome Evolution.Mol Biol Evol. 2016 Nov;33(11):3031. doi: 10.1093/molbev/msw202. Mol Biol Evol. 2016. PMID: 27738272 Free PMC article. No abstract available.
Abstract
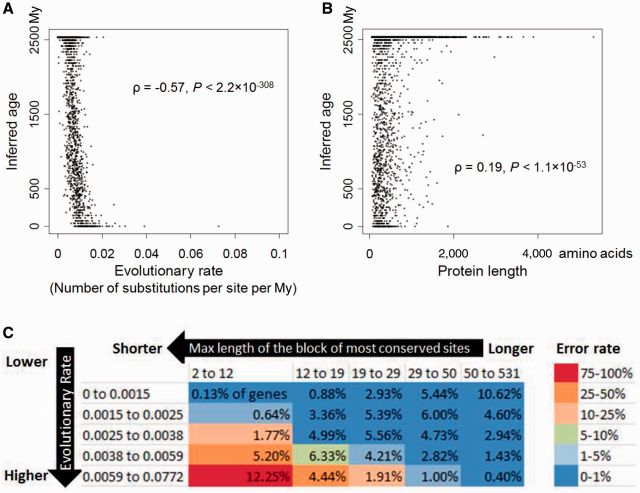
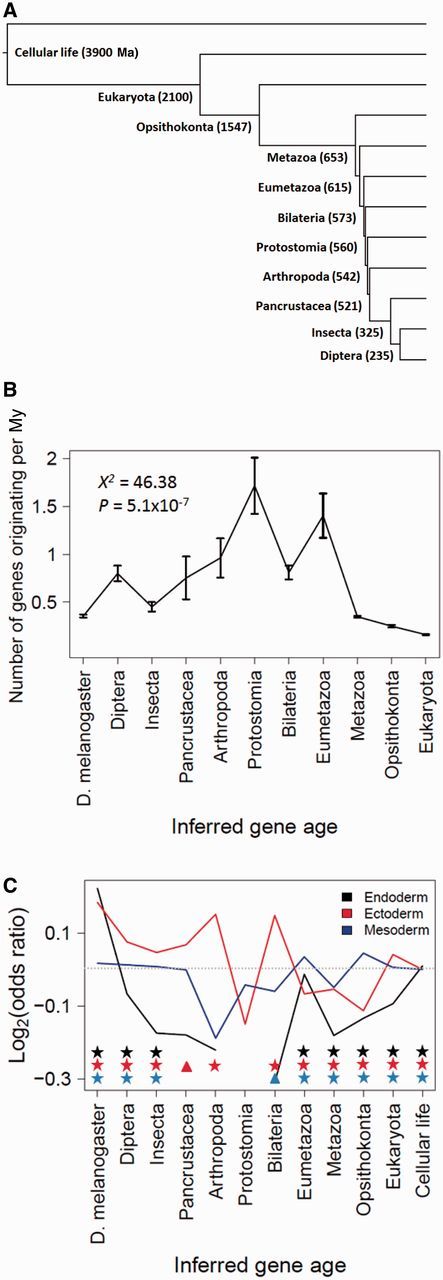
Phylostratigraphy is a method for dating the evolutionary emergence of a gene or gene family by identifying its homologs across the tree of life, typically by using BLAST searches. Applying this method to all genes in a species, or genomic phylostratigraphy, allows investigation of genome-wide patterns in new gene origination at different evolutionary times and thus has been extensively used. However, gene age estimation depends on the challenging task of detecting distant homologs via sequence similarity, which is expected to have differential accuracies for different genes. Here, we evaluate the accuracy of phylostratigraphy by realistic computer simulation with parameters estimated from genomic data, and investigate the impact of its error on findings of genome evolution. We show that 1) phylostratigraphy substantially underestimates gene age for a considerable fraction of genes, 2) the error is especially serious when the protein evolves rapidly, is short, and/or its most conserved block of sites is small, and 3) these errors create spurious nonuniform distributions of various gene properties among age groups, many of which cannot be predicted a priori. Given the high likelihood that conclusions about gene age are faulty, we advocate the use of realistic simulation to determine if observations from phylostratigraphy are explainable, at least qualitatively, by a null model of biased measurement, and in all cases, critical evaluation of results.
Keywords: BLAST; gene age; phylogenetic dating.
© The Author 2014. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures



References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
