

The genomic diversification of the whole Acinetobacter genus: origins, mechanisms, and consequences
- PMID: 25313016
- PMCID: PMC4224351
- DOI: 10.1093/gbe/evu225
The genomic diversification of the whole Acinetobacter genus: origins, mechanisms, and consequences
Abstract
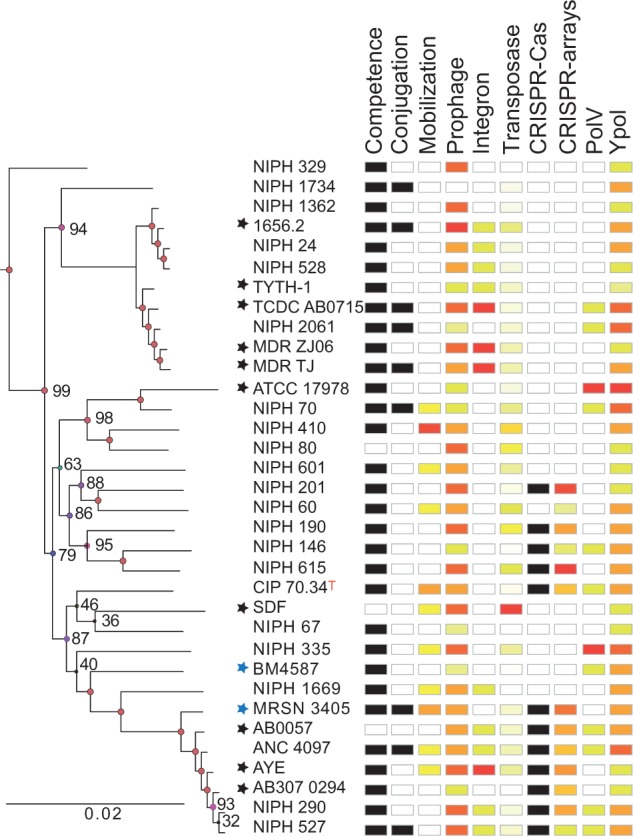
Bacterial genomics has greatly expanded our understanding of microdiversification patterns within a species, but analyses at higher taxonomical levels are necessary to understand and predict the independent rise of pathogens in a genus. We have sampled, sequenced, and assessed the diversity of genomes of validly named and tentative species of the Acinetobacter genus, a clade including major nosocomial pathogens and biotechnologically important species. We inferred a robust global phylogeny and delimited several new putative species. The genus is very ancient and extremely diverse: Genomes of highly divergent species share more orthologs than certain strains within a species. We systematically characterized elements and mechanisms driving genome diversification, such as conjugative elements, insertion sequences, and natural transformation. We found many error-prone polymerases that may play a role in resistance to toxins, antibiotics, and in the generation of genetic variation. Surprisingly, temperate phages, poorly studied in Acinetobacter, were found to account for a significant fraction of most genomes. Accordingly, many genomes encode clustered regularly interspaced short palindromic repeats (CRISPR)-Cas systems with some of the largest CRISPR-arrays found so far in bacteria. Integrons are strongly overrepresented in Acinetobacter baumannii, which correlates with its frequent resistance to antibiotics. Our data suggest that A. baumannii arose from an ancient population bottleneck followed by population expansion under strong purifying selection. The outstanding diversification of the species occurred largely by horizontal transfer, including some allelic recombination, at specific hotspots preferentially located close to the replication terminus. Our work sets a quantitative basis to understand the diversification of Acinetobacter into emerging resistant and versatile pathogens.
Keywords: bacterial genus; comparative genomics; evolution; mobile genetic elements; nosocomial pathogens.
© The Author(s) 2014. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures









References
-
- Ackermann HW, Brochu G, Emadi Konjin HP. Classification of Acinetobacter phages. Arch Virol. 1994;135:345–354. - PubMed
-
- Alvarez-Perez S, Lievens B, Jacquemyn H, Herrera CM. Acinetobacter nectaris sp. nov. and Acinetobacter boissieri sp. nov., isolated from floral nectar of wild Mediterranean insect-pollinated plants. Int J Syst Evol Microbiol. 2013;63:1532–1539. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
