

The distribution of pairwise genetic distances: a tool for investigating disease transmission
- PMID: 25313129
- PMCID: PMC4256759
- DOI: 10.1534/genetics.114.171538
The distribution of pairwise genetic distances: a tool for investigating disease transmission
Abstract
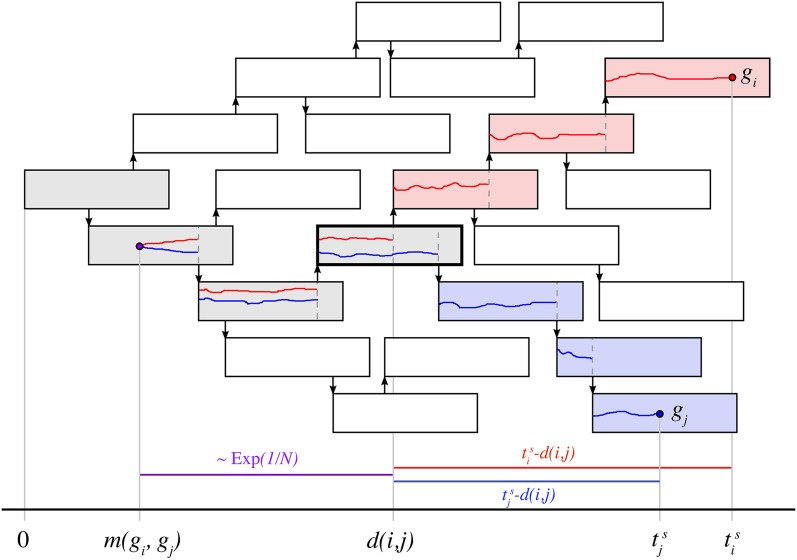
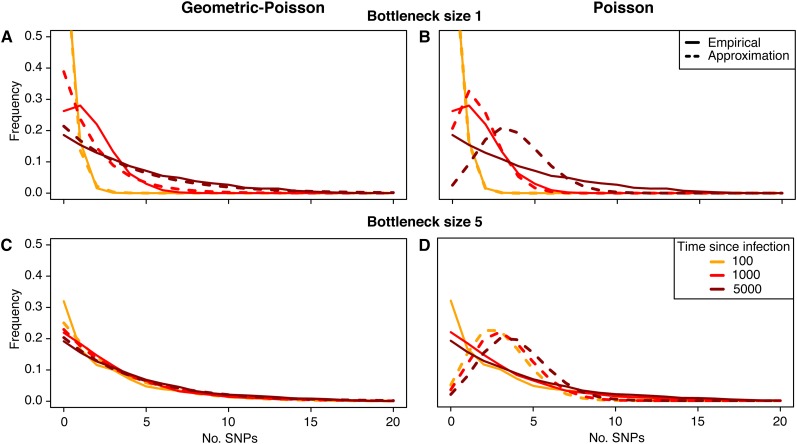
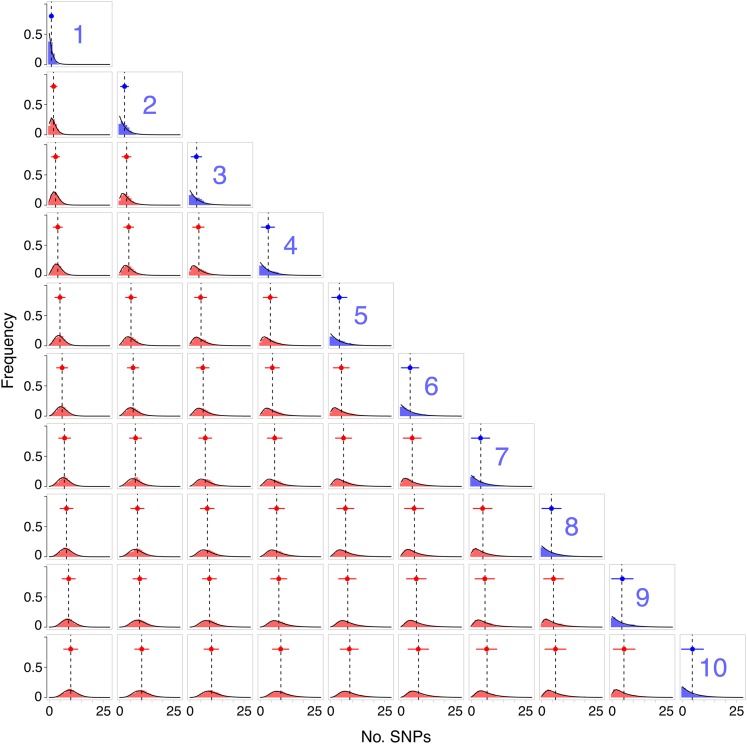
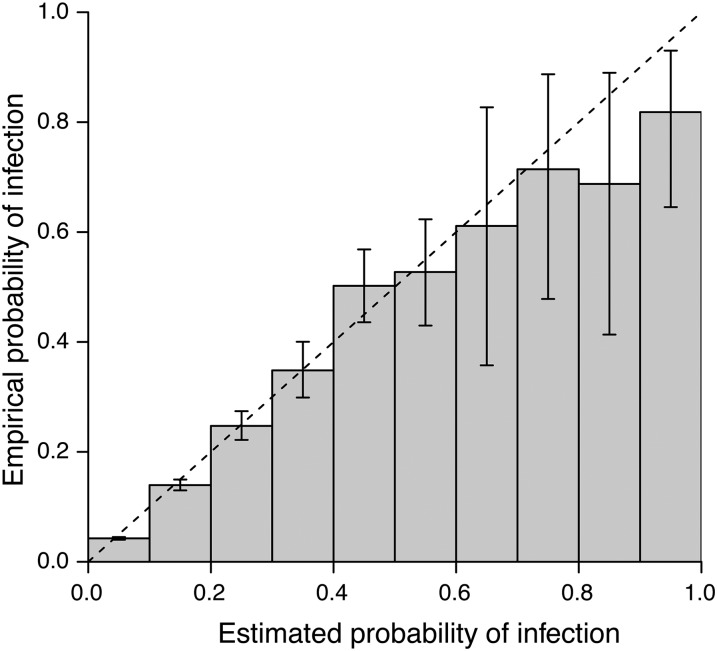
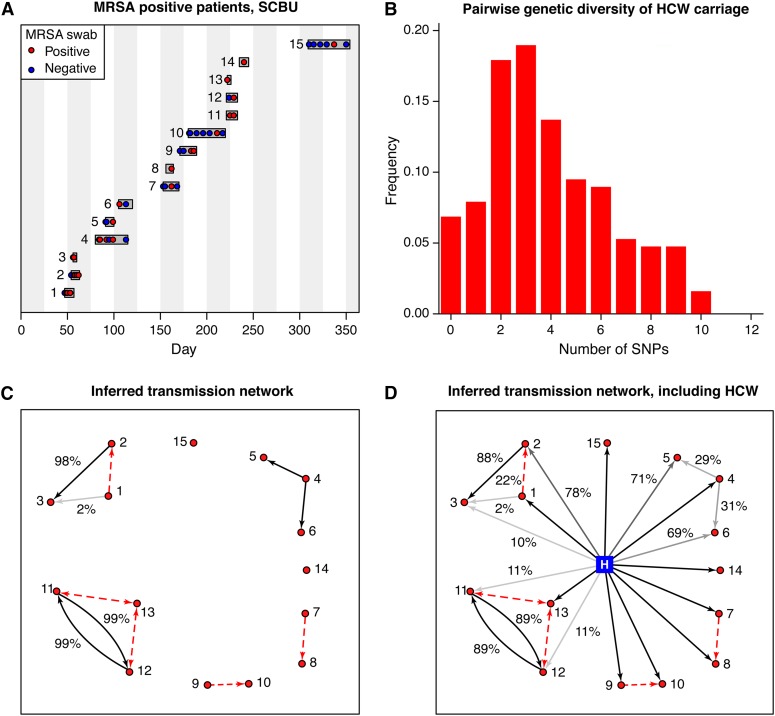
Whole-genome sequencing of pathogens has recently been used to investigate disease outbreaks and is likely to play a growing role in real-time epidemiological studies. Methods to analyze high-resolution genomic data in this context are still lacking, and inferring transmission dynamics from such data typically requires many assumptions. While recent studies have proposed methods to infer who infected whom based on genetic distance between isolates from different individuals, the link between epidemiological relationship and genetic distance is still not well understood. In this study, we investigated the distribution of pairwise genetic distances between samples taken from infected hosts during an outbreak. We proposed an analytically tractable approximation to this distribution, which provides a framework to evaluate the likelihood of particular transmission routes. Our method accounts for the transmission of a genetically diverse inoculum, a possibility overlooked in most analyses. We demonstrated that our approximation can provide a robust estimation of the posterior probability of transmission routes in an outbreak and may be used to rule out transmission events at a particular probability threshold. We applied our method to data collected during an outbreak of methicillin-resistant Staphylococcus aureus, ruling out several potential transmission links. Our study sheds light on the accumulation of mutations in a pathogen during an epidemic and provides tools to investigate transmission dynamics, avoiding the intensive computation necessary in many existing methods.
Keywords: epidemics; genetic distance; infectious diseases; transmission routes.
Copyright © 2014 by the Genetics Society of America.
Figures
References
-
- Balloux, F., 2010 Demographic influences on bacterial population structure, pp. 103–120 in Bacterial Population Genetics in Infectious Diseases, edited by D. A. Robinson, D. Falush, and E. J. Feil. John Wiley & Sons, New York.
-
- Chang-Li X., Hou-Kuhan T., Zhau-Hua S., Song-Sheng Q., Yao-Ting L., et al. , 1988. Microcalorimetric study of bacterial growth. Thermochim. Acta 123: 33–41.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
