

Evidence-based guideline summary: diagnosis and treatment of limb-girdle and distal dystrophies [RETIRED]: report of the guideline development subcommittee of the American Academy of Neurology and the practice issues review panel of the American Association of Neuromuscular & Electrodiagnostic Medicine
- PMID: 25313375
- PMCID: PMC4206155
- DOI: 10.1212/WNL.0000000000000892
Evidence-based guideline summary: diagnosis and treatment of limb-girdle and distal dystrophies [RETIRED]: report of the guideline development subcommittee of the American Academy of Neurology and the practice issues review panel of the American Association of Neuromuscular & Electrodiagnostic Medicine
Abstract
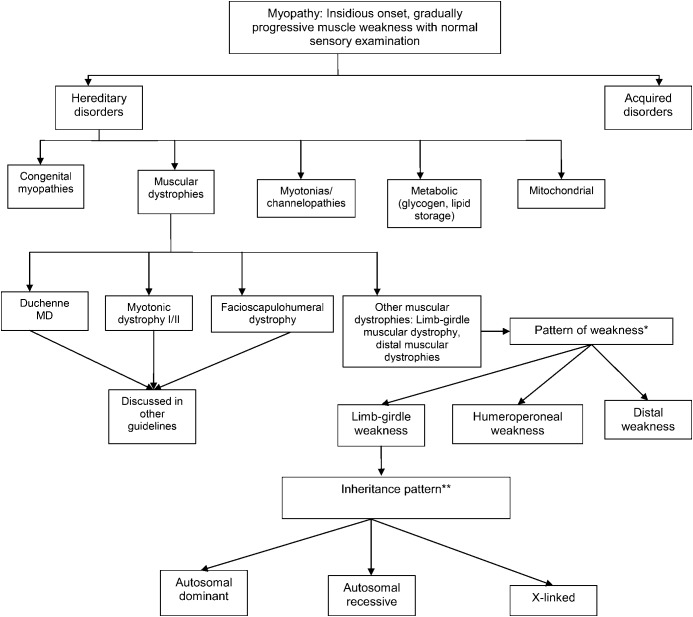
Objective: To review the current evidence and make practice recommendations regarding the diagnosis and treatment of limb-girdle muscular dystrophies (LGMDs).
Methods: Systematic review and practice recommendation development using the American Academy of Neurology guideline development process.
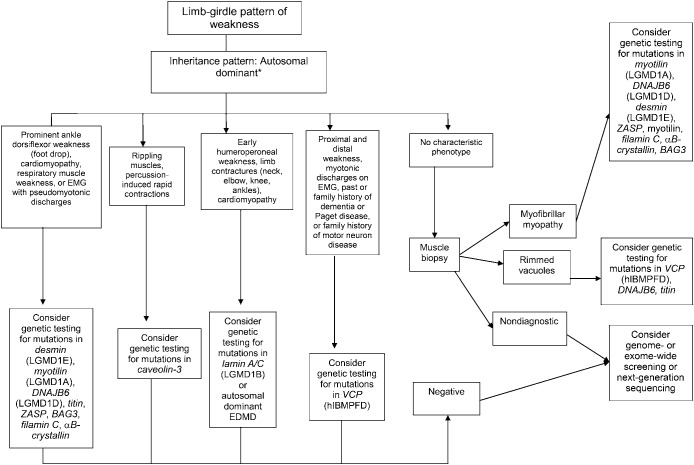
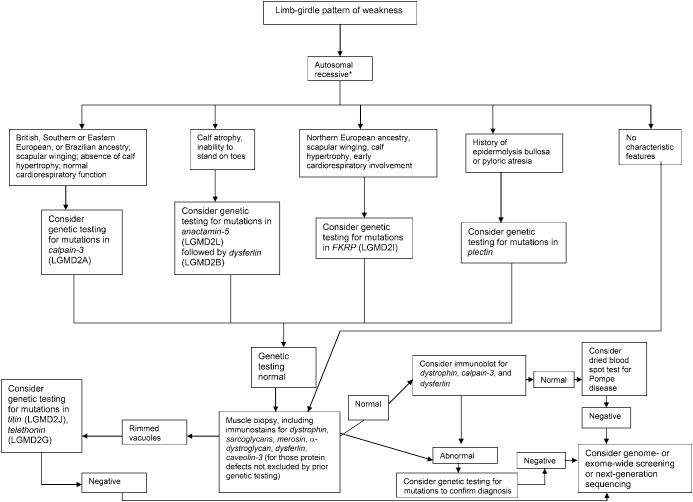
Results: Most LGMDs are rare, with estimated prevalences ranging from 0.07 per 100,000 to 0.43 per 100,000. The frequency of some muscular dystrophies varies based on the ethnic background of the population studied. Some LGMD subtypes have distinguishing features, including pattern of muscle involvement, cardiac abnormalities, extramuscular involvement, and muscle biopsy findings. The few published therapeutic trials were not designed to establish clinical efficacy of any treatment.
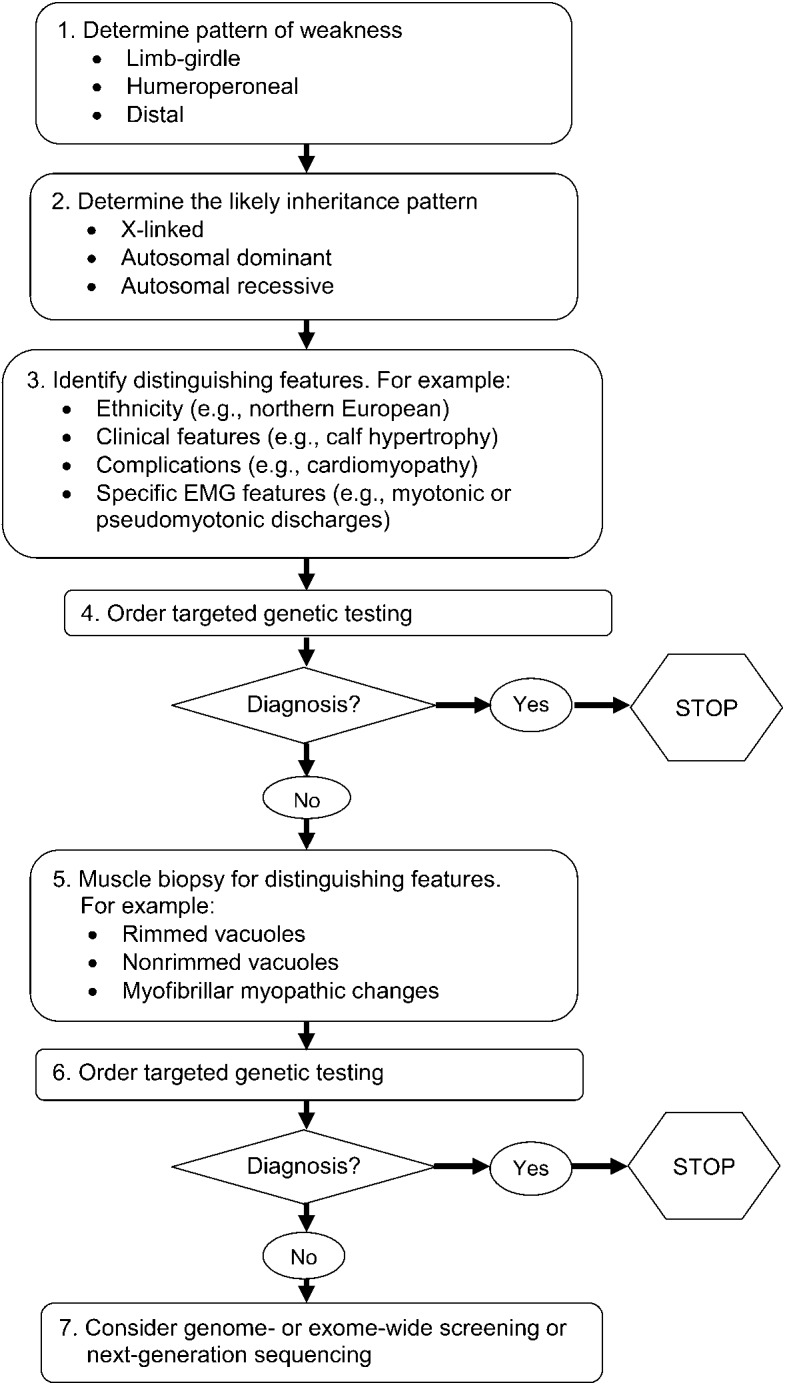
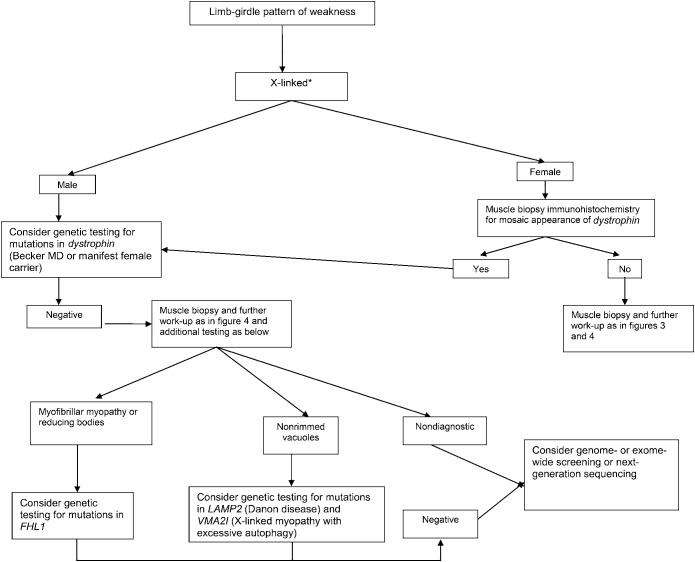
Principal recommendations: For patients with suspected muscular dystrophy, clinicians should use a clinical approach to guide genetic diagnosis based on clinical phenotype, inheritance pattern, and associated manifestations (Level B). Clinicians should refer newly diagnosed patients with an LGMD subtype and high risk of cardiac complications for cardiology evaluation even if they are asymptomatic from a cardiac standpoint (Level B). In patients with LGMD with a known high risk of respiratory failure, clinicians should obtain periodic pulmonary function testing (Level B). Clinicians should refer patients with muscular dystrophy to a clinic that has access to multiple specialties designed specifically to care for patients with neuromuscular disorders (Level B). Clinicians should not offer patients with LGMD gene therapy, myoblast transplantation, neutralizing antibody to myostatin, or growth hormone outside of a research study designed to determine efficacy and safety of the treatment (Level R). Detailed results and recommendations are available on the Neurology® Web site at Neurology.org.
© 2014 American Academy of Neurology.
Figures
Comment in
-
Evidence-based guideline summary: Diagnosis and treatment of limb-girdle and distal dystrophies: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine.Neurology. 2015 Apr 21;84(16):1720. doi: 10.1212/WNL.0000000000001508. Neurology. 2015. PMID: 25901060 No abstract available.
-
Evidence-based guideline summary: Diagnosis and treatment of limb-girdle and distal dystrophies: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine.Neurology. 2015 Apr 21;84(16):1720-1. Neurology. 2015. PMID: 26082957 No abstract available.
References
-
- Walton JN, Nattrass FJ. On the classification, natural history and treatment of the myopathies. Brain 1954;77:169–231 - PubMed
-
- Bushby KM. Diagnostic criteria for the limb-girdle muscular dystrophies: report of the ENMC Consortium on Limb-Girdle Dystrophies. Neuromuscul Disord 1995;5:71–74 - PubMed
-
- Bushby KM, Beckmann JS. The limb-girdle muscular dystrophies: proposal for a new nomenclature. Neuromuscul Disord 1995;5:337–343 - PubMed
-
- American Academy of Neurology. Guideline Development Process Manual, 2004 ed. St. Paul, MN: American Academy of Neurology; 2004
-
- American Academy of Neurology. Clinical Practice Guideline Process Manual, 2011 ed. St. Paul, MN: American Academy of Neurology; 2011
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical