

Neurologic abnormalities in mouse models of the lysosomal storage disorders mucolipidosis II and mucolipidosis III γ
- PMID: 25314316
- PMCID: PMC4196941
- DOI: 10.1371/journal.pone.0109768
Neurologic abnormalities in mouse models of the lysosomal storage disorders mucolipidosis II and mucolipidosis III γ
Erratum in
- PLoS One. 2014;9(11):e114199
Abstract
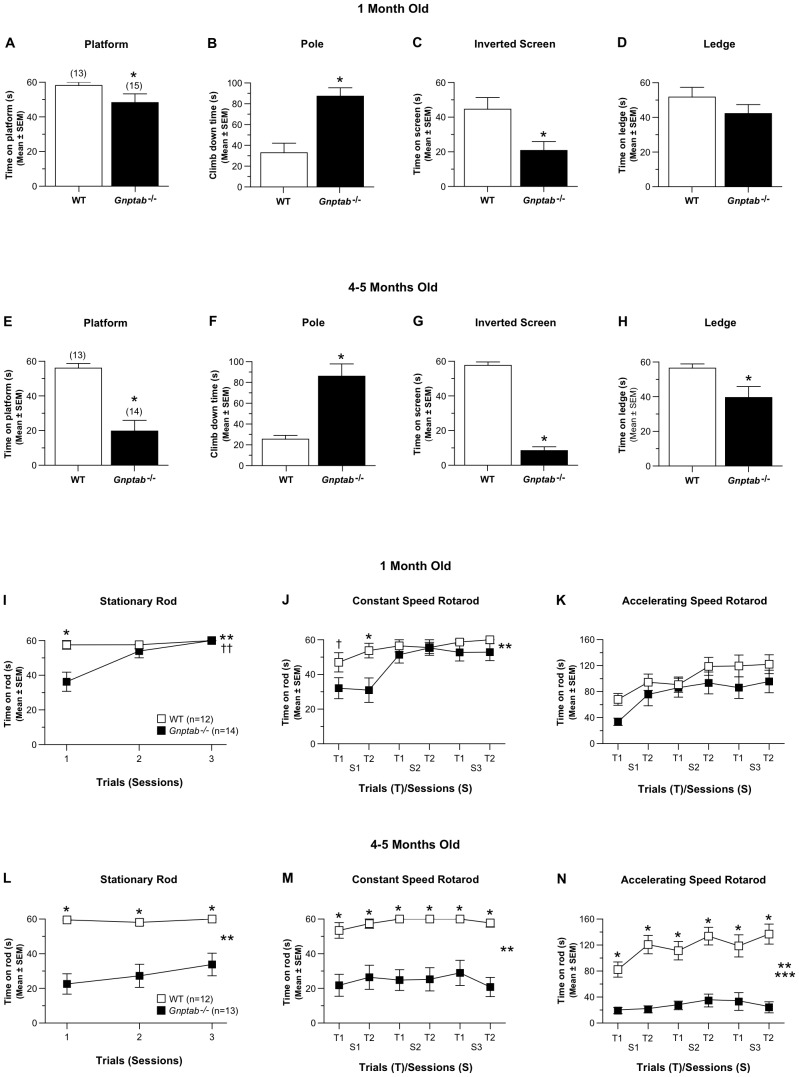
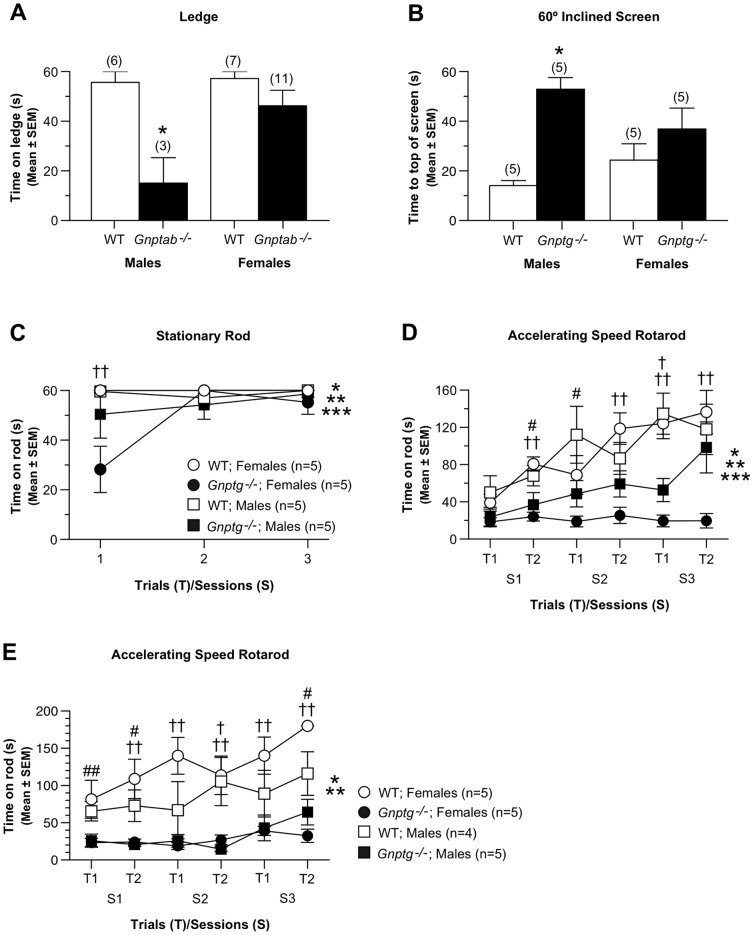
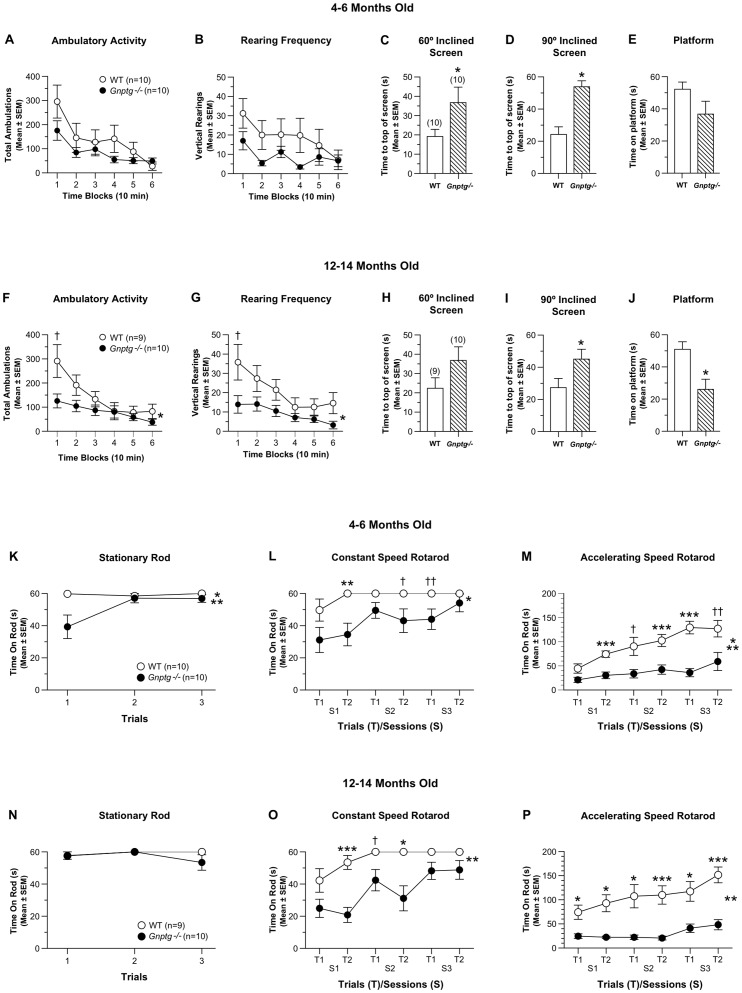
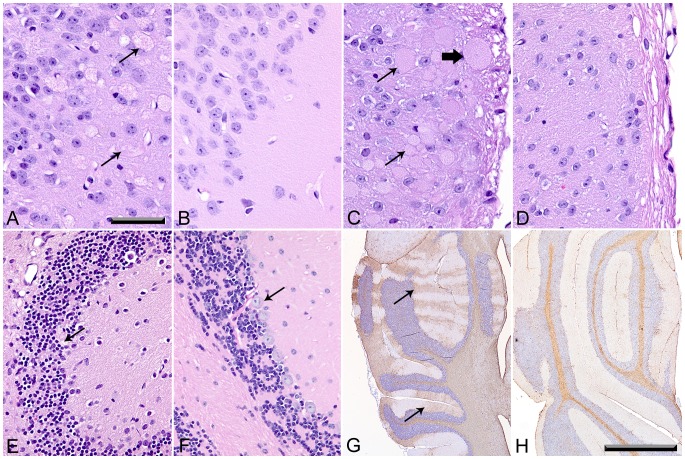
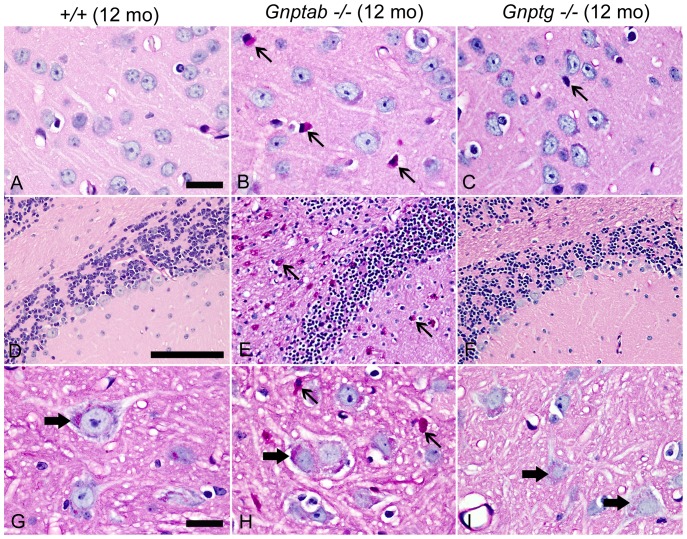
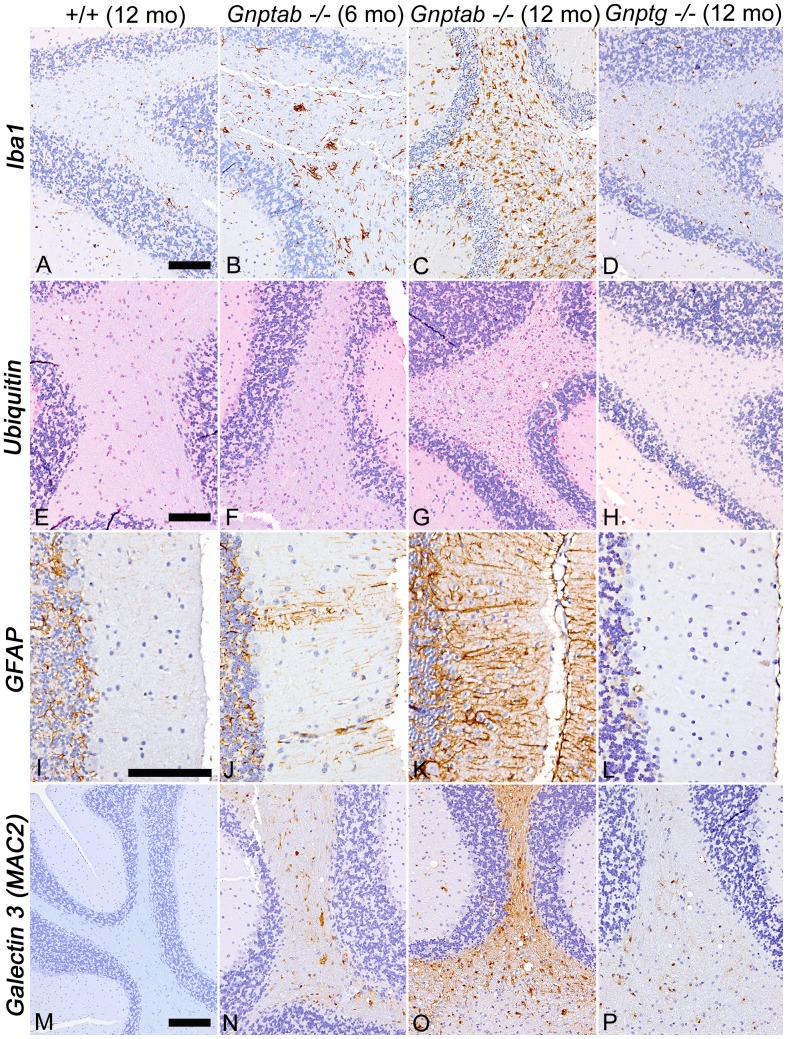
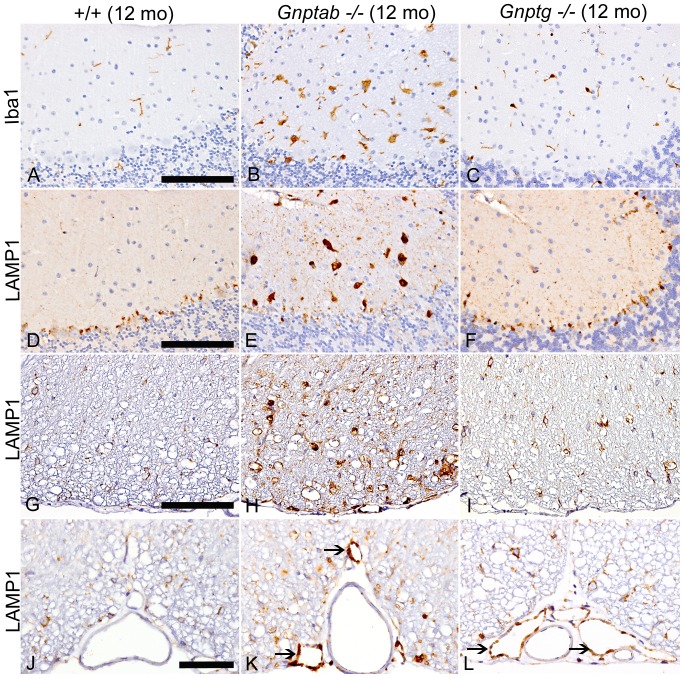
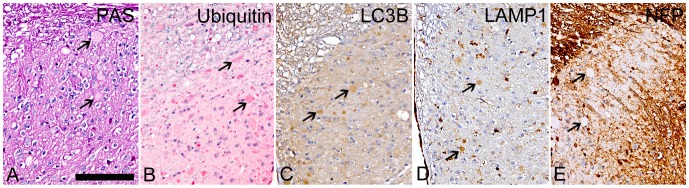
UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase is an α2β2γ2 hexameric enzyme that catalyzes the synthesis of the mannose 6-phosphate targeting signal on lysosomal hydrolases. Mutations in the α/β subunit precursor gene cause the severe lysosomal storage disorder mucolipidosis II (ML II) or the more moderate mucolipidosis III alpha/beta (ML III α/β), while mutations in the γ subunit gene cause the mildest disorder, mucolipidosis III gamma (ML III γ). Here we report neurologic consequences of mouse models of ML II and ML III γ. The ML II mice have a total loss of acid hydrolase phosphorylation, which results in depletion of acid hydrolases in mesenchymal-derived cells. The ML III γ mice retain partial phosphorylation. However, in both cases, total brain extracts have normal or near normal activity of many acid hydrolases reflecting mannose 6-phosphate-independent lysosomal targeting pathways. While behavioral deficits occur in both models, the onset of these changes occurs sooner and the severity is greater in the ML II mice. The ML II mice undergo progressive neurodegeneration with neuronal loss, astrocytosis, microgliosis and Purkinje cell depletion which was evident at 4 months whereas ML III γ mice have only mild to moderate astrocytosis and microgliosis at 12 months. Both models accumulate the ganglioside GM2, but only ML II mice accumulate fucosylated glycans. We conclude that in spite of active mannose 6-phosphate-independent targeting pathways in the brain, there are cell types that require at least partial phosphorylation function to avoid lysosomal dysfunction and the associated neurodegeneration and behavioral impairments.
Conflict of interest statement
Figures
References
-
- Braulke T, Raas-Rothschild A, Kornfeld S (2014) I-Cell Disease and Pseudo-Hurler Polydystrophy: Disorders of Lysosomal Enzyme Phosphorylation and Localization. In: Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A et al.., editors. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: The McGraw-Hill Companies, Inc.
-
- Cathey SS, Kudo M, Tiede S, Raas-Rothschild A, Braulke T, et al. (2008) Molecular order in mucolipidosis II and III nomenclature. American journal of medical genetics Part A 146A: 512–513. - PubMed
-
- Dittmer F, Ulbrich EJ, Hafner A, Schmahl W, Meister T, et al. (1999) Alternative mechanisms for trafficking of lysosomal enzymes in mannose 6-phosphate receptor-deficient mice are cell type-specific. Journal of cell science 112 (Pt 10): 1591–1597. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
