

Viewing protein fitness landscapes through a next-gen lens
- PMID: 25316787
- PMCID: PMC4196605
- DOI: 10.1534/genetics.114.168351
Viewing protein fitness landscapes through a next-gen lens
Abstract
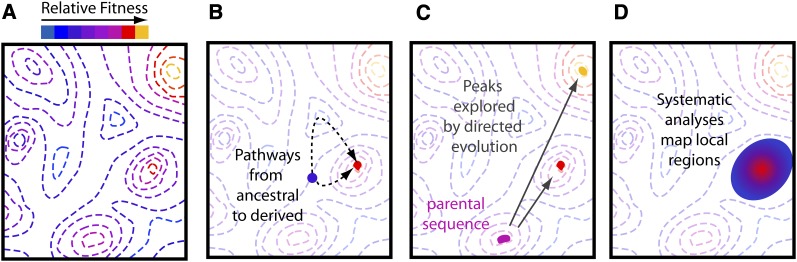
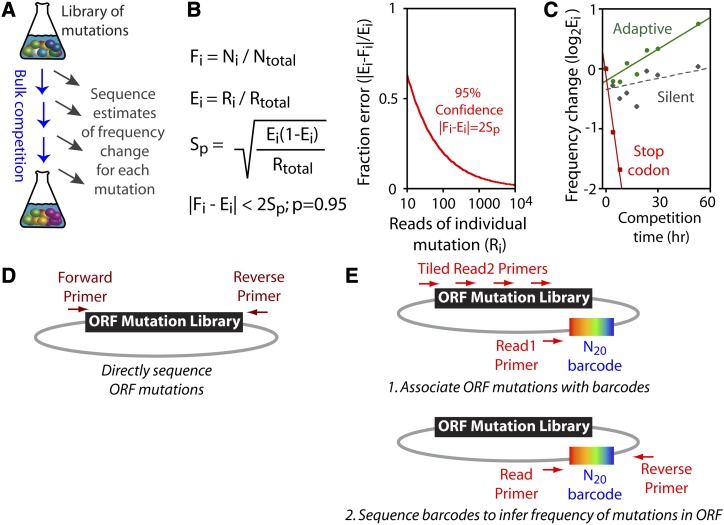
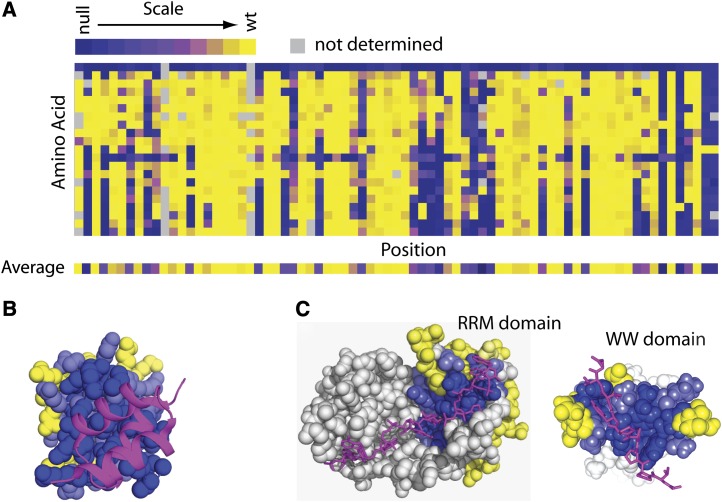
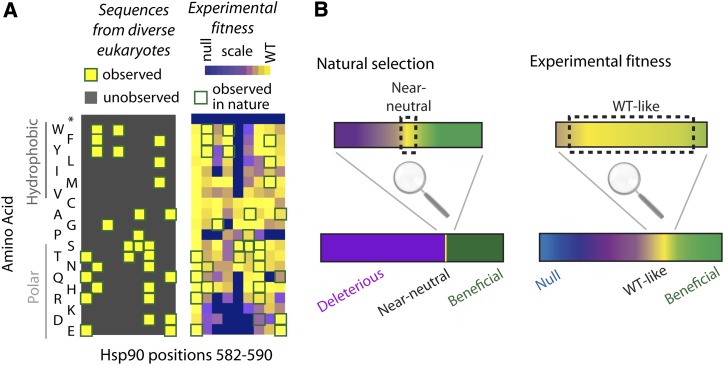
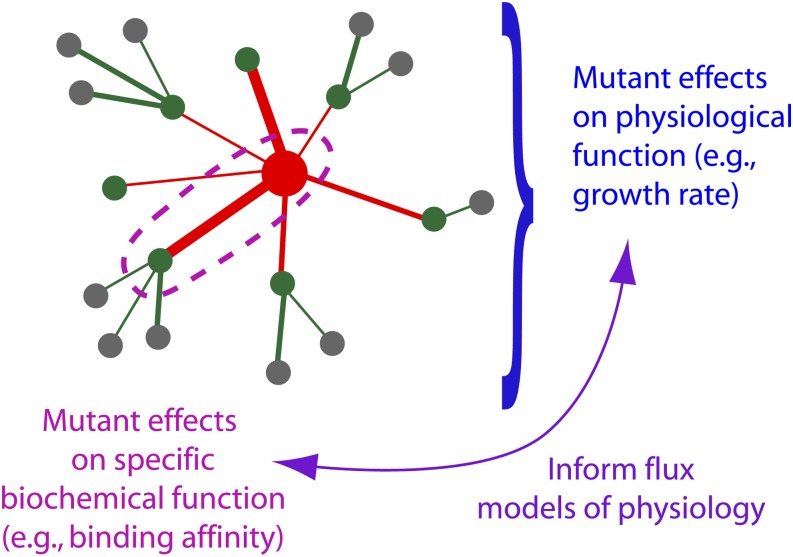
High-throughput sequencing has enabled many powerful approaches in biological research. Here, we review sequencing approaches to measure frequency changes within engineered mutational libraries subject to selection. These analyses can provide direct estimates of biochemical and fitness effects for all individual mutations across entire genes (and likely compact genomes in the near future) in genetically tractable systems such as microbes, viruses, and mammalian cells. The effects of mutations on experimental fitness can be assessed using sequencing to monitor time-dependent changes in mutant frequency during bulk competitions. The impact of mutations on biochemical functions can be determined using reporters or other means of separating variants based on individual activities (e.g., binding affinity for a partner molecule can be interrogated using surface display of libraries of mutant proteins and isolation of bound and unbound populations). The comprehensive investigation of mutant effects on both biochemical function and experimental fitness provide promising new avenues to investigate the connections between biochemistry, cell physiology, and evolution. We summarize recent findings from systematic mutational analyses; describe how they relate to a field rich in both theory and experimentation; and highlight how they may contribute to ongoing and future research into protein structure-function relationships, systems-level descriptions of cell physiology, and population-genetic inferences on the relative contributions of selection and drift.
Keywords: mutant effects; physiology; protein function; systems biology.
Copyright © 2014 by the Genetics Society of America.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
