

Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense
- PMID: 25316792
- PMCID: PMC4217423
- DOI: 10.1073/pnas.1412767111
Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense
Abstract
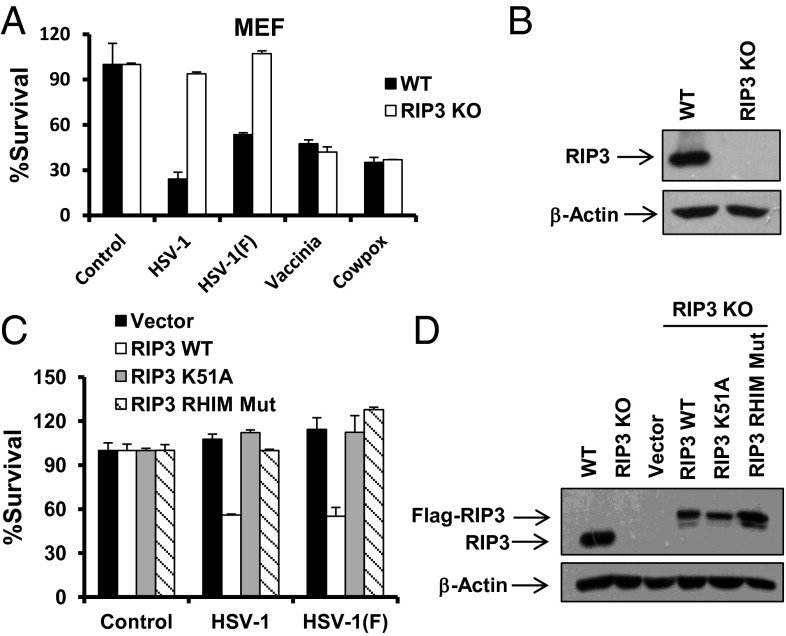
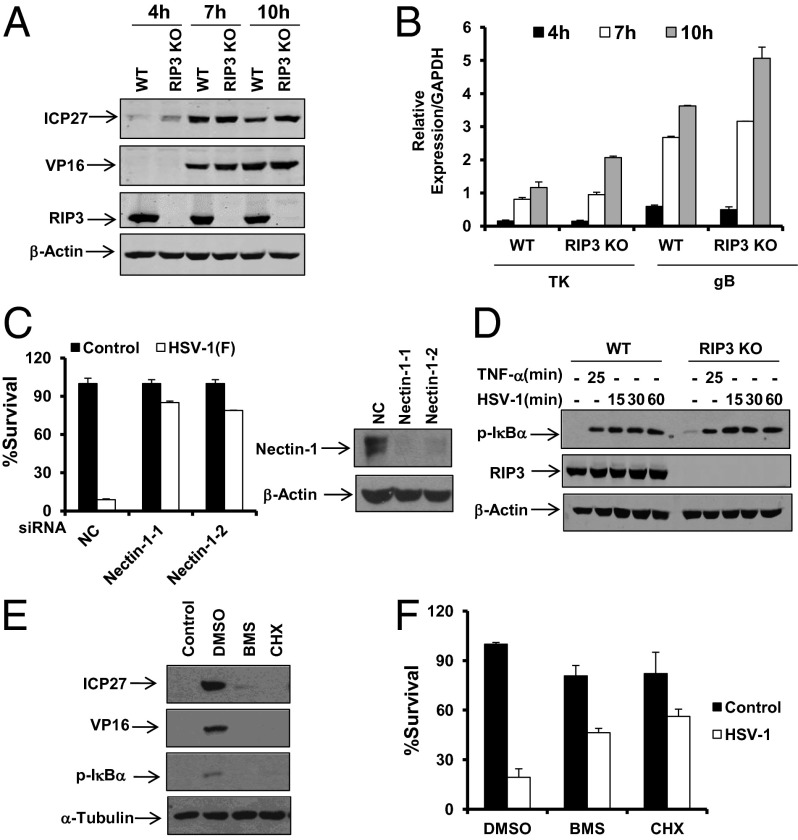
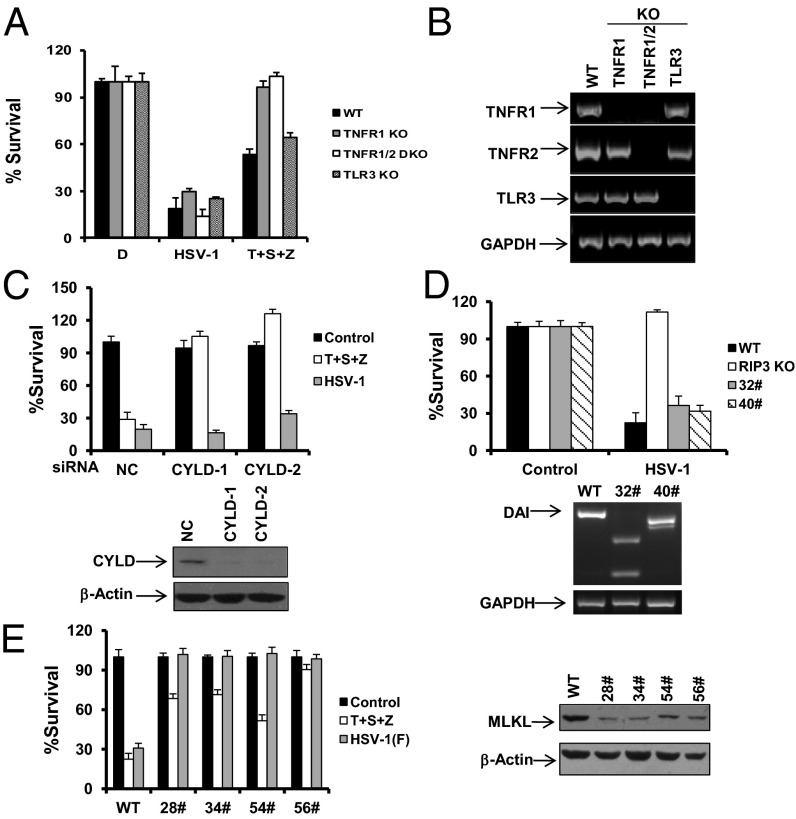
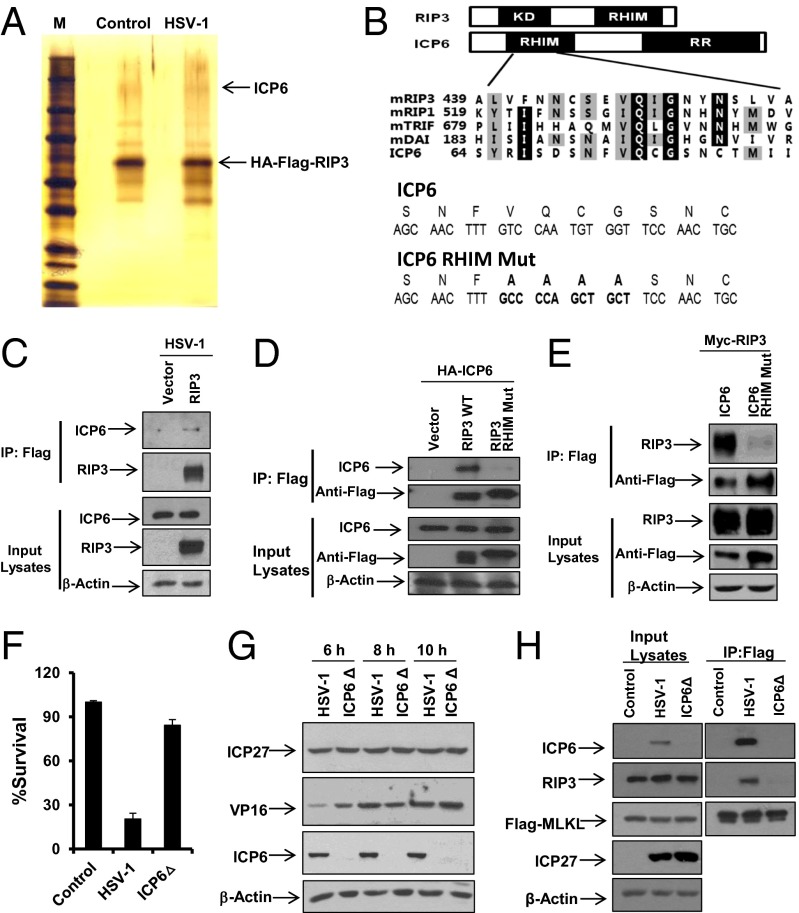
The receptor-interacting kinase-3 (RIP3) and its downstream substrate mixed lineage kinase domain-like protein (MLKL) have emerged as the key cellular components in programmed necrotic cell death. Receptors for the cytokines of tumor necrosis factor (TNF) family and Toll-like receptors (TLR) 3 and 4 are able to activate RIP3 through receptor-interacting kinase-1 and Toll/IL-1 receptor domain-containing adapter inducing IFN-β, respectively. This form of cell death has been implicated in the host-defense system. However, the molecular mechanisms that drive the activation of RIP3 by a variety of pathogens, other than the above-mentioned receptors, are largely unknown. Here, we report that human herpes simplex virus 1 (HSV-1) infection triggers RIP3-dependent necrosis. This process requires MLKL but is independent of TNF receptor, TLR3, cylindromatosis, and host RIP homotypic interaction motif-containing protein DNA-dependent activator of IFN regulatory factor. After HSV-1 infection, the viral ribonucleotide reductase large subunit (ICP6) interacts with RIP3. The formation of the ICP6-RIP3 complex requires the RHIM domains of both proteins. An HSV-1 ICP6 deletion mutant failed to cause effective necrosis of HSV-1-infected cells. Furthermore, ectopic expression of ICP6, but not RHIM mutant ICP6, directly activated RIP3/MLKL-mediated necrosis. Mice lacking RIP3 exhibited severely impaired control of HSV-1 replication and pathogenesis. Therefore, this study reveals a previously uncharacterized host antipathogen mechanism.
Keywords: HSV-1; ICP6; MLKL; RIP3; programmed necrosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
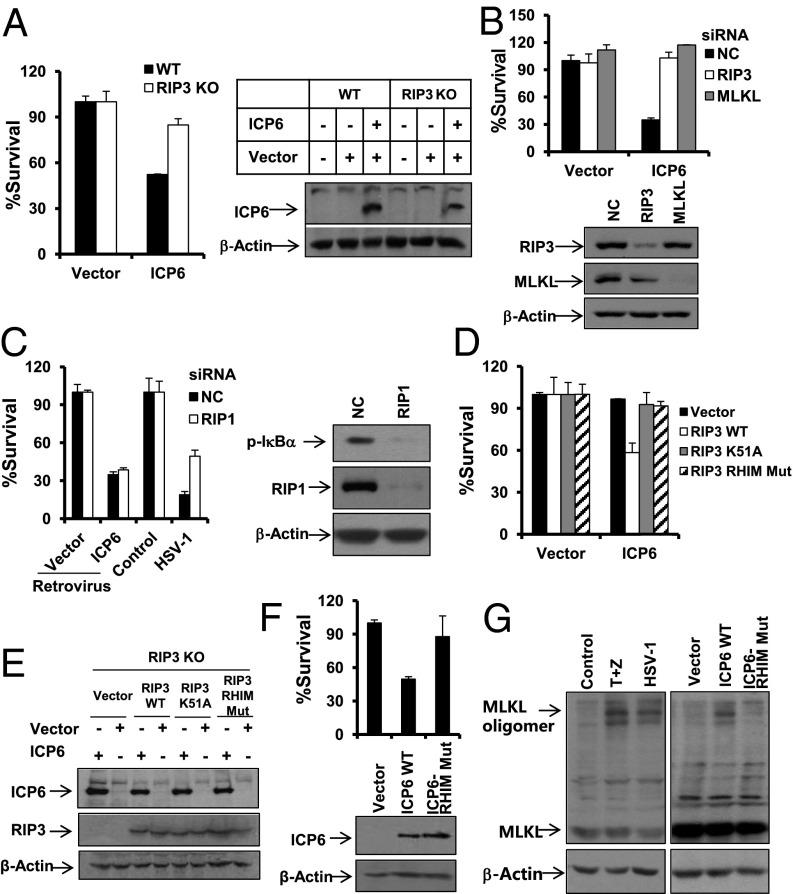
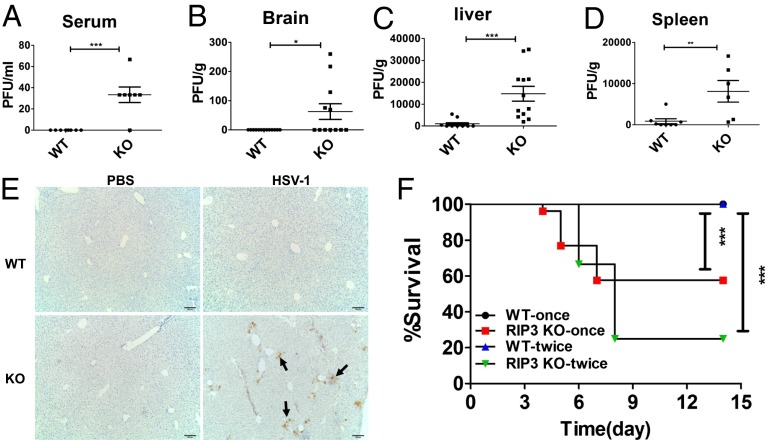
Comment in
-
HSV cheats the executioner.Cell Host Microbe. 2015 Feb 11;17(2):148-51. doi: 10.1016/j.chom.2015.01.013. Cell Host Microbe. 2015. PMID: 25674981
References
-
- Thornberry NA, Lazebnik Y. Caspases: Enemies within. Science. 1998;281(5381):1312–1316. - PubMed
-
- Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 2010;8(1):44–54. - PubMed
-
- Han J, Zhong CQ, Zhang DW. Programmed necrosis: Backup to and competitor with apoptosis in the immune system. Nat Immunol. 2011;12(12):1143–1149. - PubMed
-
- Degterev A, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
