

Origins of major archaeal clades correspond to gene acquisitions from bacteria
- PMID: 25317564
- PMCID: PMC4285555
- DOI: 10.1038/nature13805
Origins of major archaeal clades correspond to gene acquisitions from bacteria
Abstract
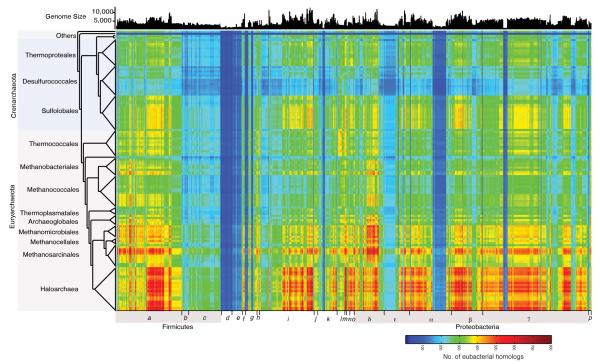
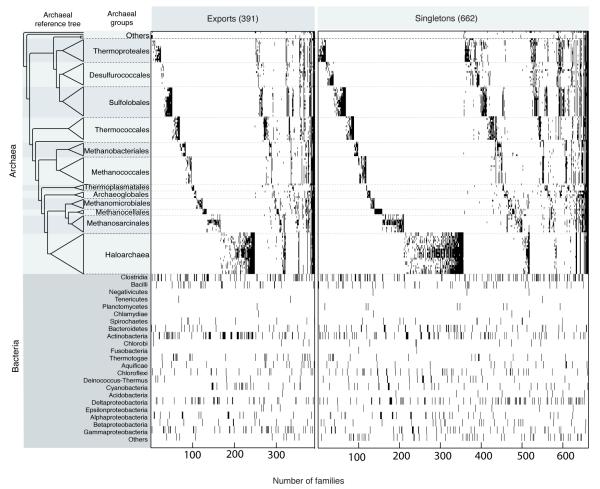
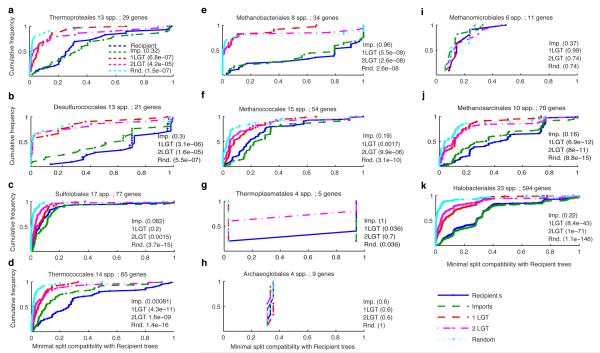
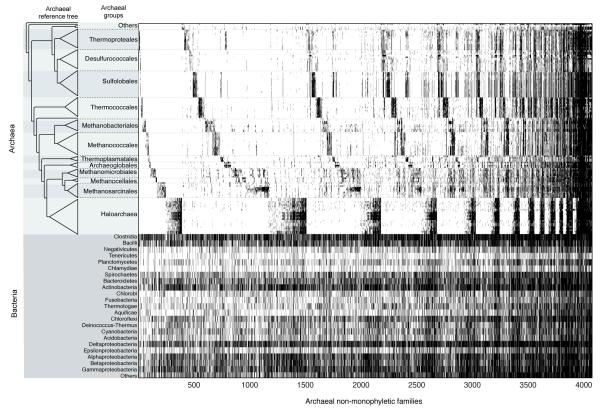
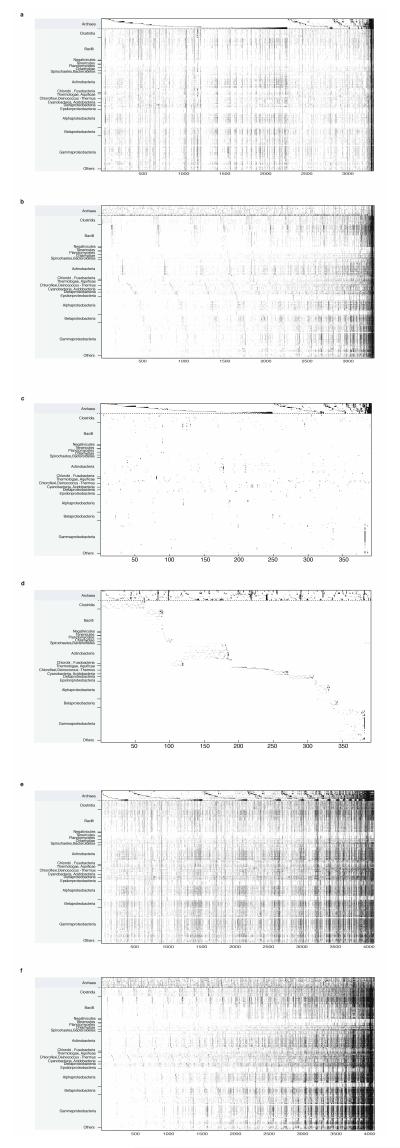
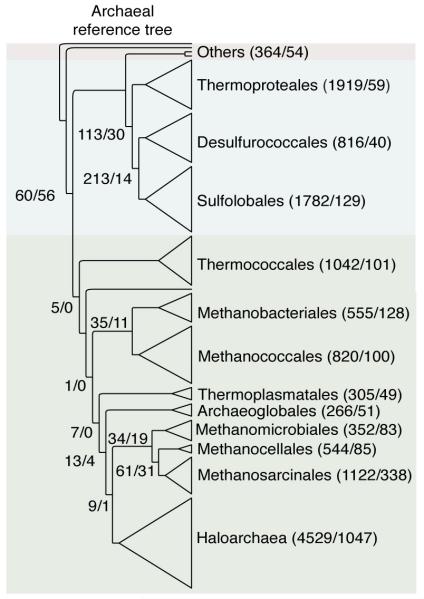
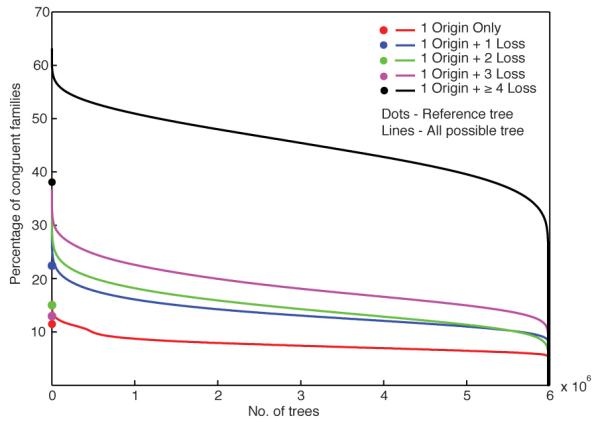
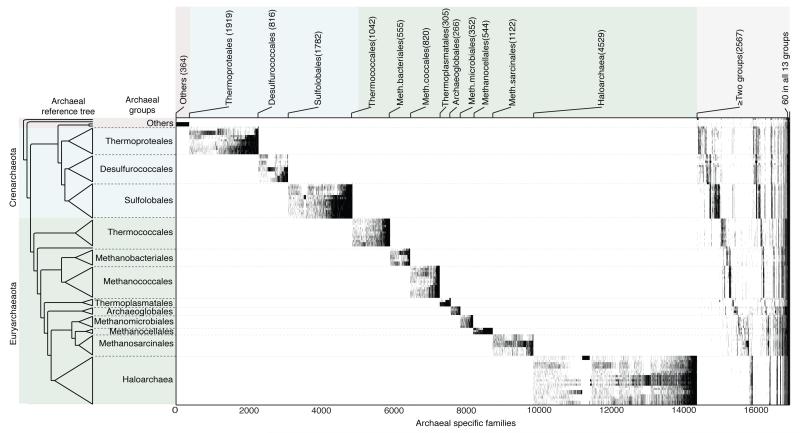
The mechanisms that underlie the origin of major prokaryotic groups are poorly understood. In principle, the origin of both species and higher taxa among prokaryotes should entail similar mechanisms--ecological interactions with the environment paired with natural genetic variation involving lineage-specific gene innovations and lineage-specific gene acquisitions. To investigate the origin of higher taxa in archaea, we have determined gene distributions and gene phylogenies for the 267,568 protein-coding genes of 134 sequenced archaeal genomes in the context of their homologues from 1,847 reference bacterial genomes. Archaeal-specific gene families define 13 traditionally recognized archaeal higher taxa in our sample. Here we report that the origins of these 13 groups unexpectedly correspond to 2,264 group-specific gene acquisitions from bacteria. Interdomain gene transfer is highly asymmetric, transfers from bacteria to archaea are more than fivefold more frequent than vice versa. Gene transfers identified at major evolutionary transitions among prokaryotes specifically implicate gene acquisitions for metabolic functions from bacteria as key innovations in the origin of higher archaeal taxa.
Figures

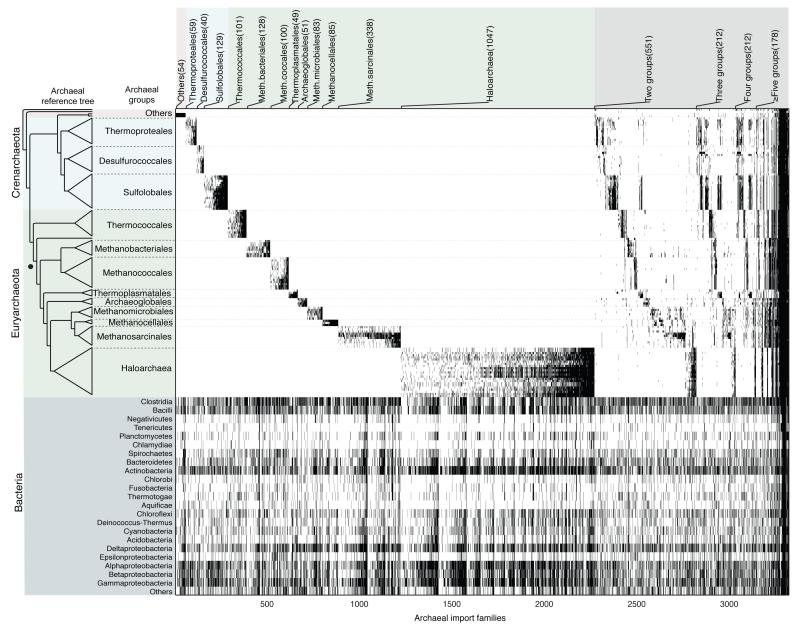
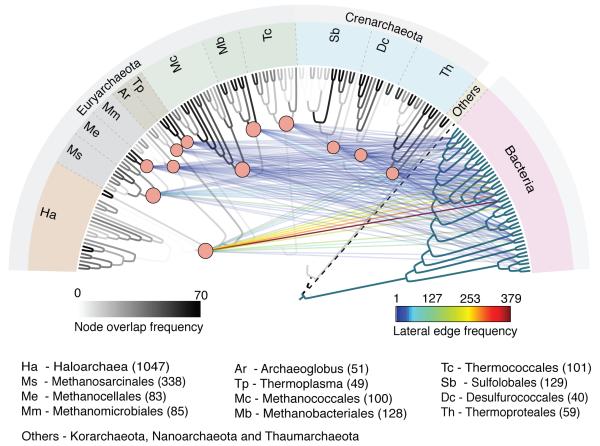
Comment in
-
Metabolic bacterial genes and the construction of high-level composite lineages of life.Trends Ecol Evol. 2015 Mar;30(3):127-9. doi: 10.1016/j.tree.2015.01.001. Epub 2015 Jan 16. Trends Ecol Evol. 2015. PMID: 25601290 Free PMC article.
References
-
- Retchless AC, Lawrence JG. Temporal fragmentation of speciation in Bacteria. Science. 2007;317:1093–1096. - PubMed
-
- Achtmann M, Wagner M. Microbial diversity and the genetic nature of microbial species. Nat. Rev. Microbiol. 2008;6:431–440. - PubMed
-
- Fraser C, Alm EJ, Polz MF, Spratt BG, Hanage WP. The bacterial species challenge: making sense of genetic and ecological diversity. Science. 2009;323:741–746. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
