

Dynamic architecture of a protein kinase
- PMID: 25319261
- PMCID: PMC4217441
- DOI: 10.1073/pnas.1418402111
Dynamic architecture of a protein kinase
Erratum in
- Proc Natl Acad Sci U S A. 2014 Nov 25;111(47):16973
Abstract
Protein kinases are dynamically regulated signaling proteins that act as switches in the cell by phosphorylating target proteins. To establish a framework for analyzing linkages between structure, function, dynamics, and allostery in protein kinases, we carried out multiple microsecond-scale molecular-dynamics simulations of protein kinase A (PKA), an exemplar active kinase. We identified residue-residue correlated motions based on the concept of mutual information and used the Girvan-Newman method to partition PKA into structurally contiguous "communities." Most of these communities included 40-60 residues and were associated with a particular protein kinase function or a regulatory mechanism, and well-known motifs based on sequence and secondary structure were often split into different communities. The observed community maps were sensitive to the presence of different ligands and provide a new framework for interpreting long-distance allosteric coupling. Communication between different communities was also in agreement with the previously defined architecture of the protein kinase core based on the "hydrophobic spine" network. This finding gives us confidence in suggesting that community analyses can be used for other protein kinases and will provide an efficient tool for structural biologists. The communities also allow us to think about allosteric consequences of mutations that are linked to disease.
Keywords: community analysis; molecular dynamics; phosphorylation.
Conflict of interest statement
Conflict of interest statement: M.K.G has an equity interest in, and is a cofounder and scientific advisor of, VeraChem LLC.
Figures
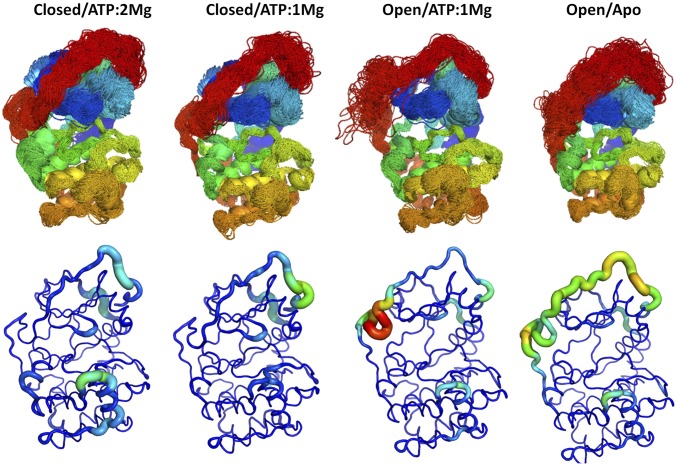
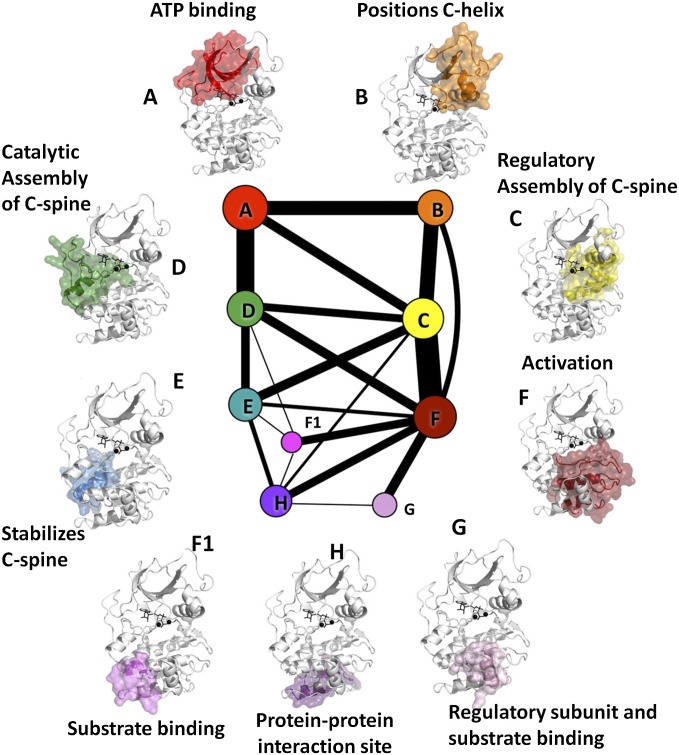
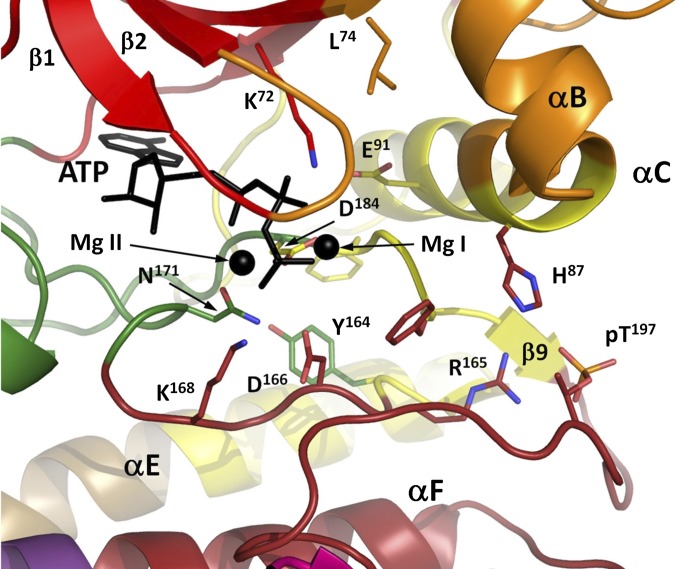
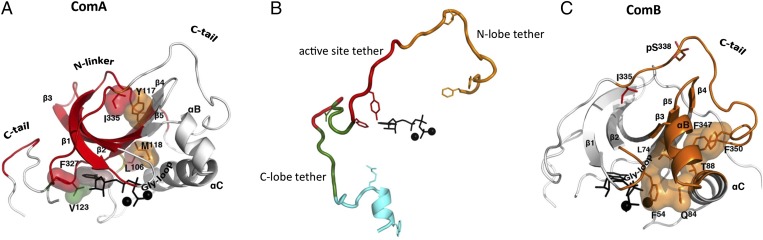
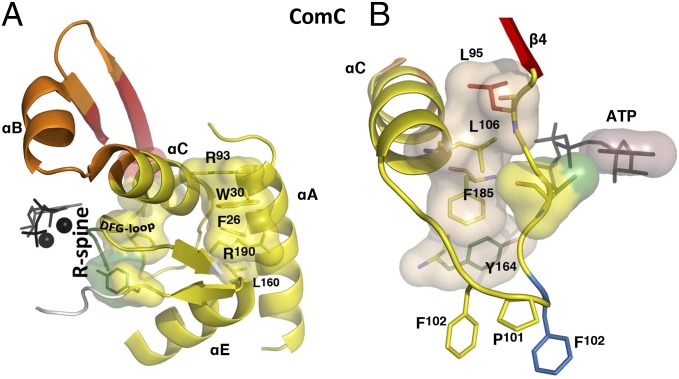
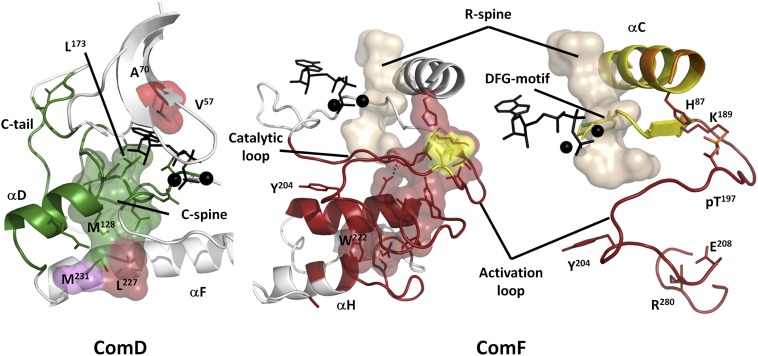
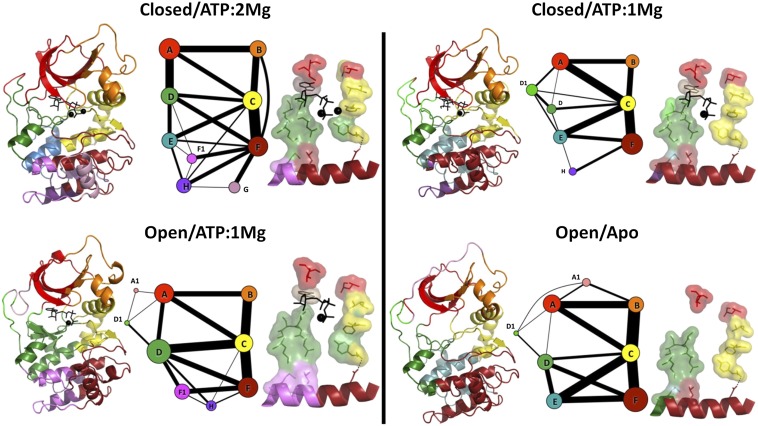
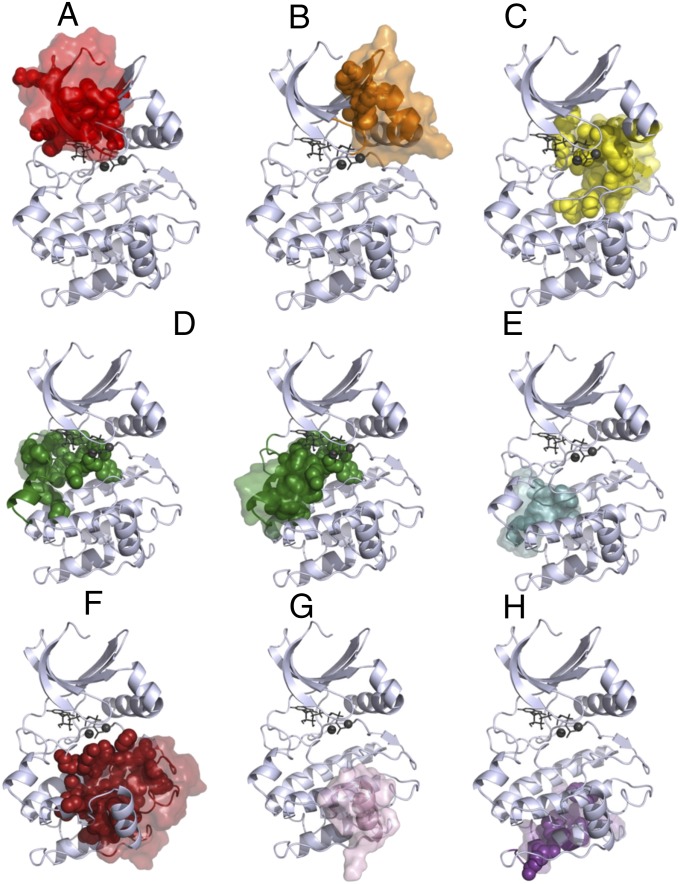
References
-
- Johnson DA, Akamine P, Radzio-Andzelm E, Madhusudan M, Taylor SS. Dynamics of cAMP-dependent protein kinase. Chem Rev. 2001;101(8):2243–2270. - PubMed
-
- Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109(3):275–282. - PubMed
-
- Hanks SK, Quinn AM, Hunter T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241(4861):42–52. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
