

Invited review: decoding the pathophysiological mechanisms that underlie RNA dysregulation in neurodegenerative disorders: a review of the current state of the art
- PMID: 25319671
- PMCID: PMC4329338
- DOI: 10.1111/nan.12187
Invited review: decoding the pathophysiological mechanisms that underlie RNA dysregulation in neurodegenerative disorders: a review of the current state of the art
Abstract
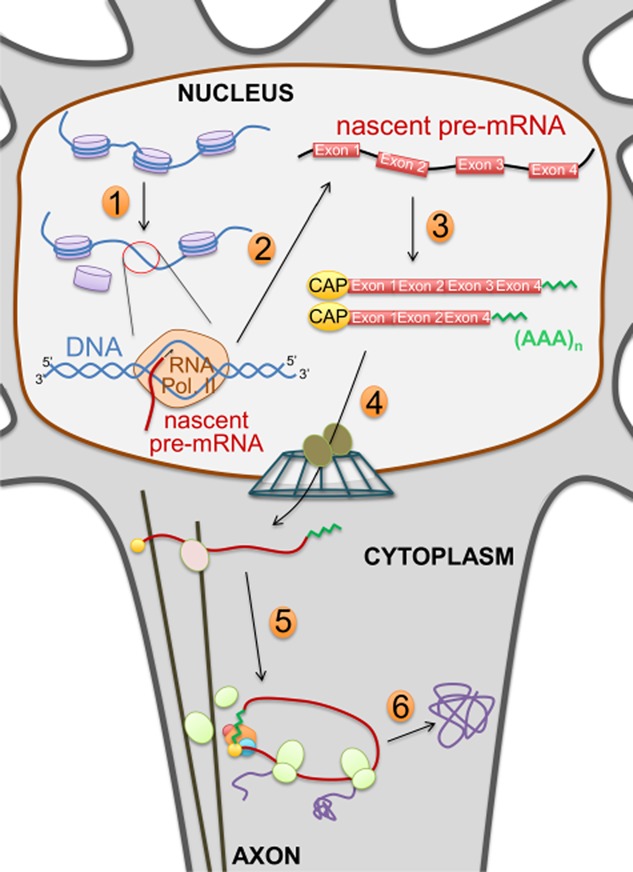
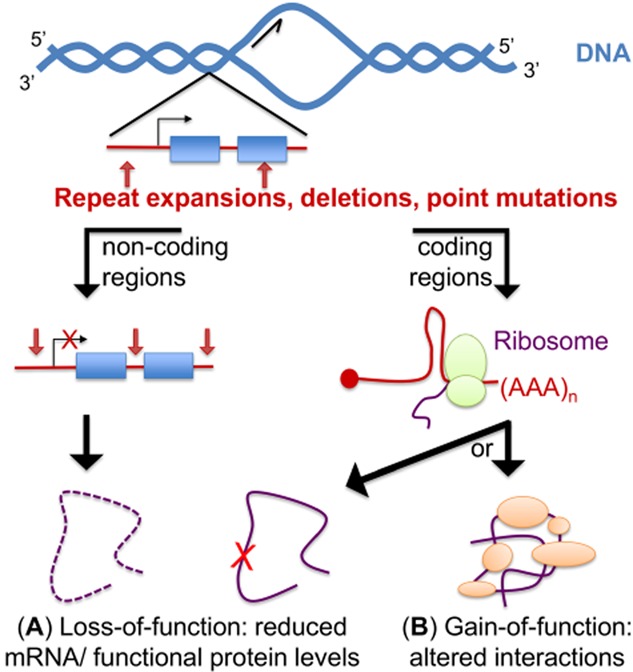
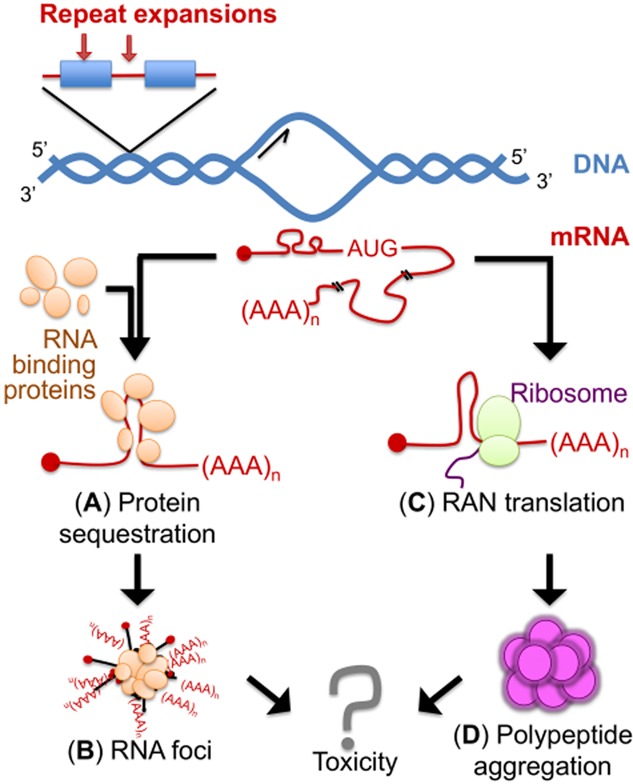
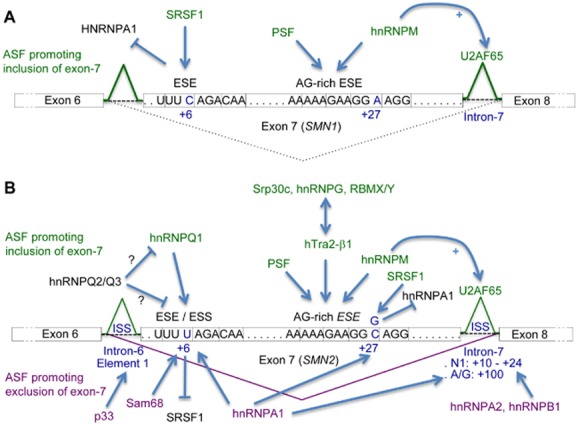
Altered RNA metabolism is a key pathophysiological component causing several neurodegenerative diseases. Genetic mutations causing neurodegeneration occur in coding and noncoding regions of seemingly unrelated genes whose products do not always contribute to the gene expression process. Several pathogenic mechanisms may coexist within a single neuronal cell, including RNA/protein toxic gain-of-function and/or protein loss-of-function. Genetic mutations that cause neurodegenerative disorders disrupt healthy gene expression at diverse levels, from chromatin remodelling, transcription, splicing, through to axonal transport and repeat-associated non-ATG (RAN) translation. We address neurodegeneration in repeat expansion disorders [Huntington's disease, spinocerebellar ataxias, C9ORF72-related amyotrophic lateral sclerosis (ALS)] and in diseases caused by deletions or point mutations (spinal muscular atrophy, most subtypes of familial ALS). Some neurodegenerative disorders exhibit broad dysregulation of gene expression with the synthesis of hundreds to thousands of abnormal messenger RNA (mRNA) molecules. However, the number and identity of aberrant mRNAs that are translated into proteins - and how these lead to neurodegeneration - remain unknown. The field of RNA biology research faces the challenge of identifying pathophysiological events of dysregulated gene expression. In conclusion, we discuss current research limitations and future directions to improve our characterization of pathological mechanisms that trigger disease onset and progression.
Keywords: RNA-mediated diseases; RNA/protein toxic gain-of-function; altered gene expression; neurodegeneration; protein loss-of-function.
© 2014 The Authors. Neuropathology and Applied Neurobiology published by John Wiley & Sons Ltd on behalf of British Neuropathological Society.
Figures
References
-
- Walker FO. Huntington's disease. Lancet. 2007;369:218–228. - PubMed
-
- MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, MacFarlane H, Jenkins B, Anderson MA, Wexler NS, Gusella JF, Bates GP, Baxendale S, Hummerich H, Kirby S, North M, Youngman S, Mott R, Zehetner G, Sedlacek Z, Poustka A, Frischauf AM, Lehrach H, Buckler AJ, Church D, Doucettestamm L, Odonovan MC, Ribaramirez L, Shah M, Stanton VP, Strobel SA, Draths KM, Wales JL, Dervan P, Housman DE, Altherr M, Shiang R, Thompson L, Fielder T, Wasmuth JJ, Tagle D, Valdes J, Elmer L, Allard M, Castilla L, Swaroop M, Blanchard K, Collins FS, Snell R, Holloway T, Gillespie K, Datson N, Shaw D, Harper PS. A novel gene containing a trinucleotide repeat that is expanded and unstable on huntingtons-disease chromosomes. Cell. 1993;72:971–983. - PubMed
-
- LaSpada AR, Paulson HL, Fischbeck KH. Trinucleotide repeat expansion in neurological disease. Ann Neurol. 1994;36:814–822. - PubMed
-
- Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–2133. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
