

Divergent functions of hematopoietic transcription factors in lineage priming and differentiation during erythro-megakaryopoiesis
- PMID: 25319996
- PMCID: PMC4248311
- DOI: 10.1101/gr.164178.113
Divergent functions of hematopoietic transcription factors in lineage priming and differentiation during erythro-megakaryopoiesis
Abstract
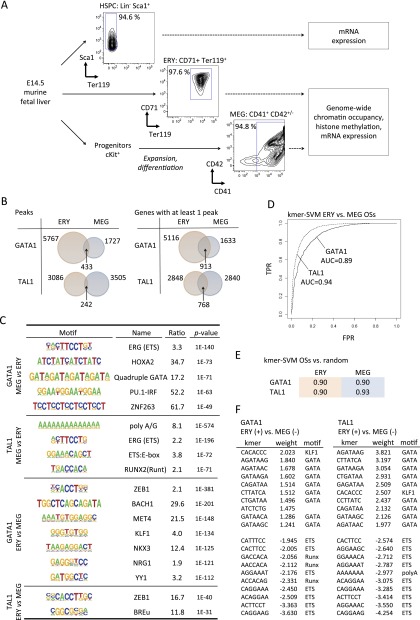
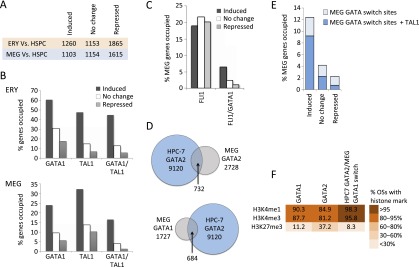
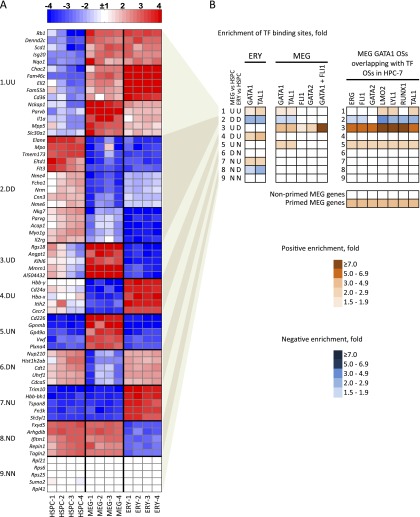
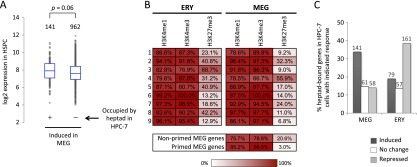
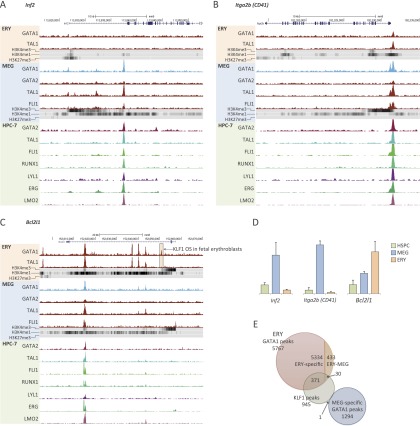
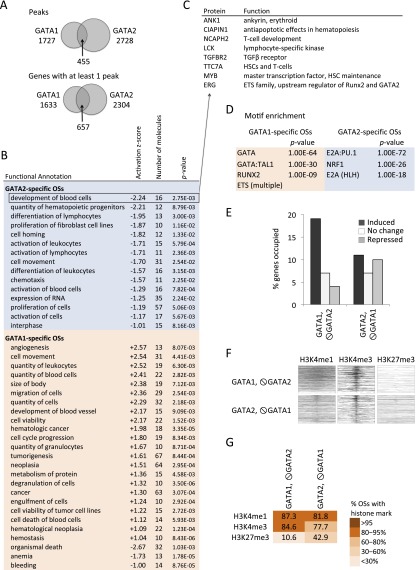
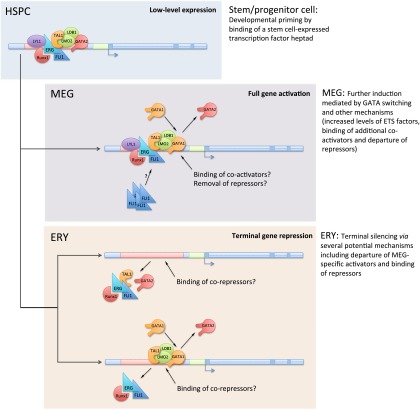
Combinatorial actions of relatively few transcription factors control hematopoietic differentiation. To investigate this process in erythro-megakaryopoiesis, we correlated the genome-wide chromatin occupancy signatures of four master hematopoietic transcription factors (GATA1, GATA2, TAL1, and FLI1) and three diagnostic histone modification marks with the gene expression changes that occur during development of primary cultured megakaryocytes (MEG) and primary erythroblasts (ERY) from murine fetal liver hematopoietic stem/progenitor cells. We identified a robust, genome-wide mechanism of MEG-specific lineage priming by a previously described stem/progenitor cell-expressed transcription factor heptad (GATA2, LYL1, TAL1, FLI1, ERG, RUNX1, LMO2) binding to MEG-associated cis-regulatory modules (CRMs) in multipotential progenitors. This is followed by genome-wide GATA factor switching that mediates further induction of MEG-specific genes following lineage commitment. Interaction between GATA and ETS factors appears to be a key determinant of these processes. In contrast, ERY-specific lineage priming is biased toward GATA2-independent mechanisms. In addition to its role in MEG lineage priming, GATA2 plays an extensive role in late megakaryopoiesis as a transcriptional repressor at loci defined by a specific DNA signature. Our findings reveal important new insights into how ERY and MEG lineages arise from a common bipotential progenitor via overlapping and divergent functions of shared hematopoietic transcription factors.
© 2014 Pimkin et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Bouilloux F, Juban G, Cohet N, Buet D, Guyot B, Vainchenker W, Louache F, Morlé F. 2008. EKLF restricts megakaryocytic differentiation at the benefit of erythrocytic differentiation. Blood 112: 576–584. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
- U01 HL099656/HL/NHLBI NIH HHS/United States
- P30 DK090969/DK/NIDDK NIH HHS/United States
- R37DK058044/DK/NIDDK NIH HHS/United States
- U01HL099656/HL/NHLBI NIH HHS/United States
- R37 DK058044/DK/NIDDK NIH HHS/United States
- U01 HL099993/HL/NHLBI NIH HHS/United States
- R01DK58044/DK/NIDDK NIH HHS/United States
- U54 HG006998/HG/NHGRI NIH HHS/United States
- R01DK54937/DK/NIDDK NIH HHS/United States
- RC2 HG005573/HG/NHGRI NIH HHS/United States
- R01 DK058044/DK/NIDDK NIH HHS/United States
- P30 CA010815/CA/NCI NIH HHS/United States
- R56 DK065806/DK/NIDDK NIH HHS/United States
- P01 HL064190/HL/NHLBI NIH HHS/United States
- 1R01HG007348-01/HG/NHGRI NIH HHS/United States
- P30DK090969/DK/NIDDK NIH HHS/United States
- R01 DK065806/DK/NIDDK NIH HHS/United States
- RC2HG005573/HG/NHGRI NIH HHS/United States
- U54HG006998/HG/NHGRI NIH HHS/United States
- R01 HG007348/HG/NHGRI NIH HHS/United States
- R01 DK054937/DK/NIDDK NIH HHS/United States
- R01DK065806/DK/NIDDK NIH HHS/United States
- P01 HL110860/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous