

A single mechanism can explain network-wide insulin resistance in adipocytes from obese patients with type 2 diabetes
- PMID: 25320095
- PMCID: PMC4246081
- DOI: 10.1074/jbc.M114.608927
A single mechanism can explain network-wide insulin resistance in adipocytes from obese patients with type 2 diabetes
Abstract
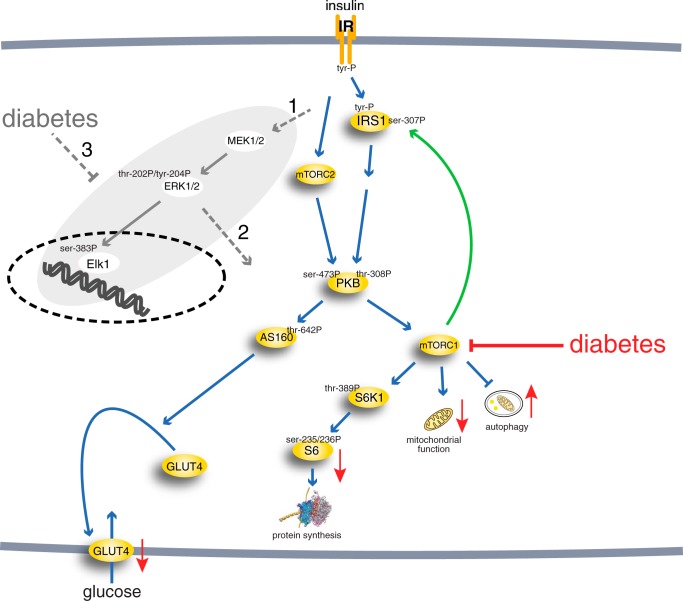
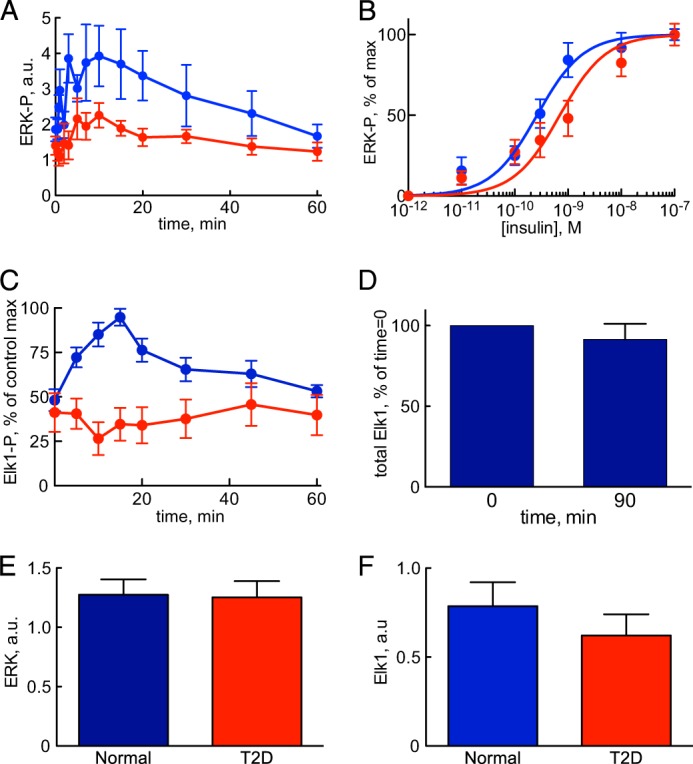
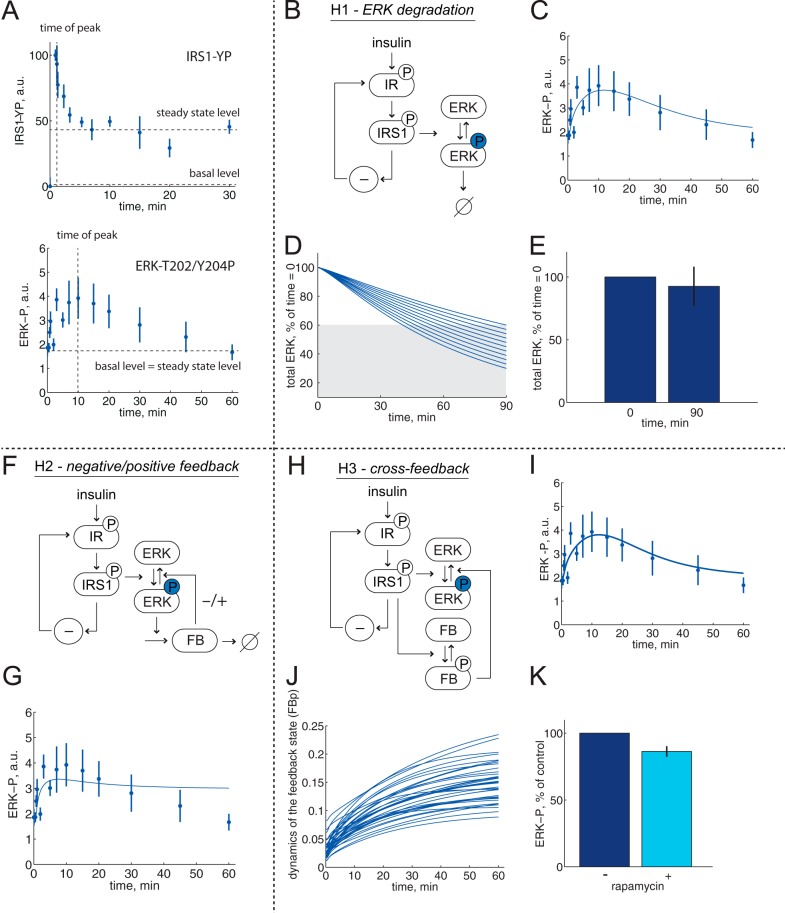
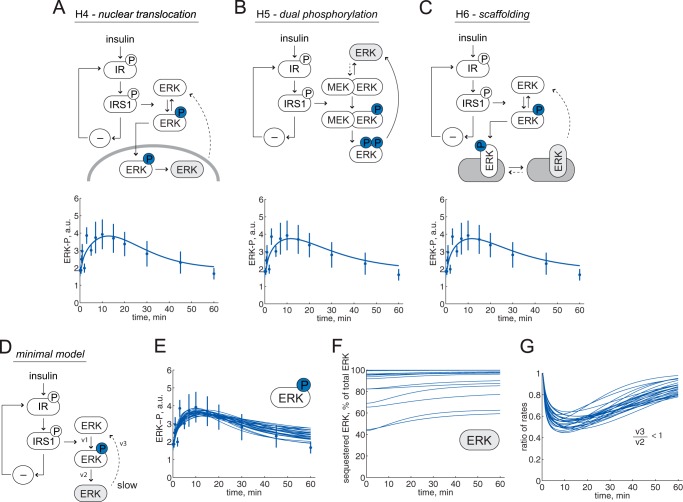
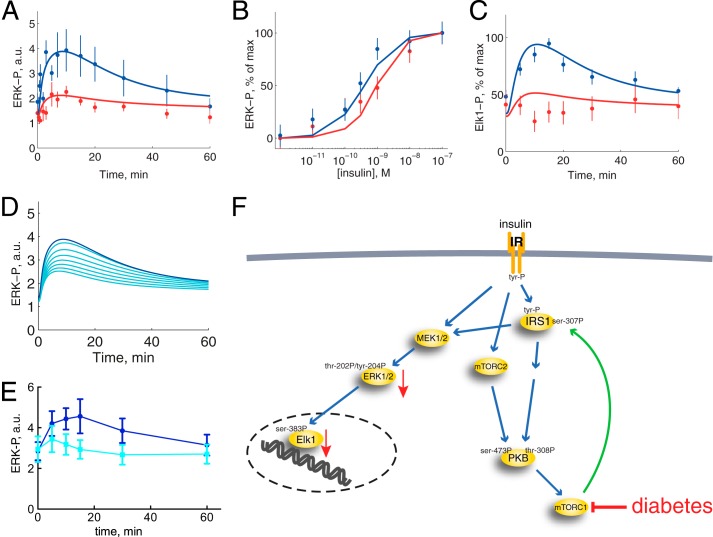
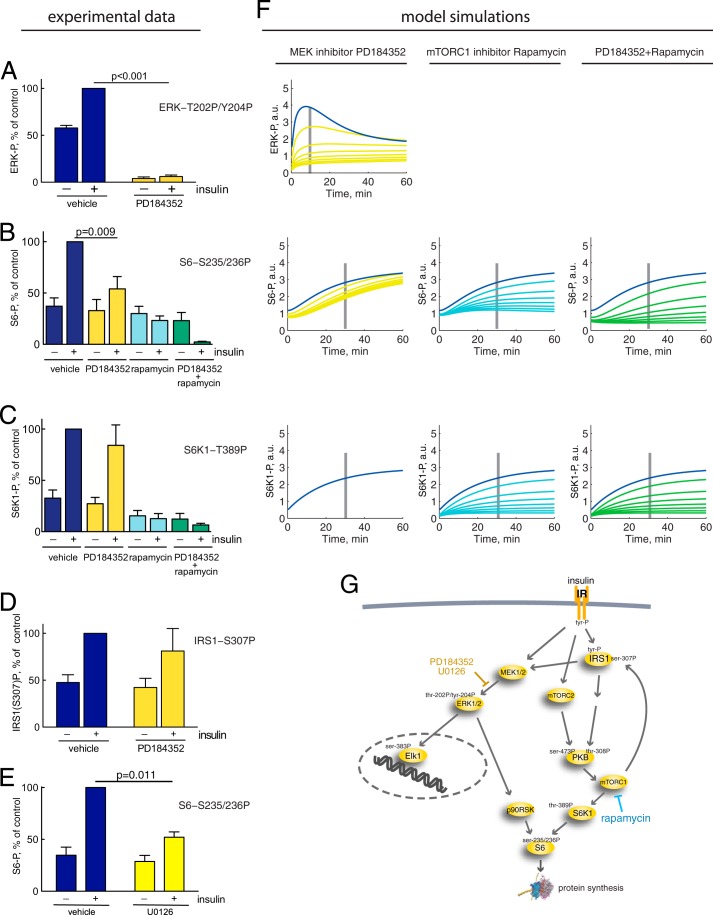
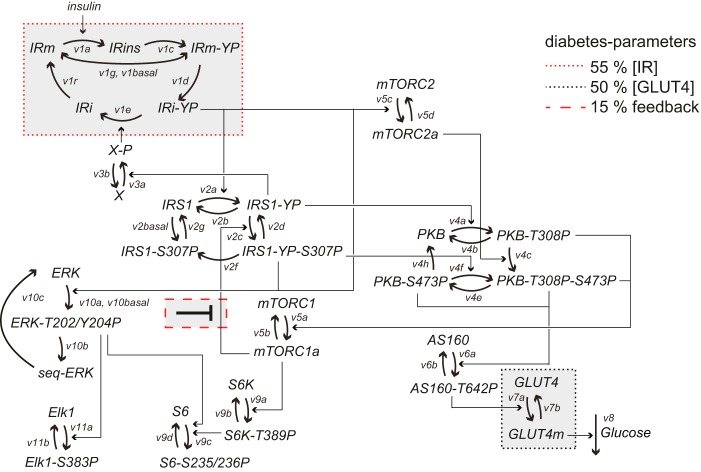
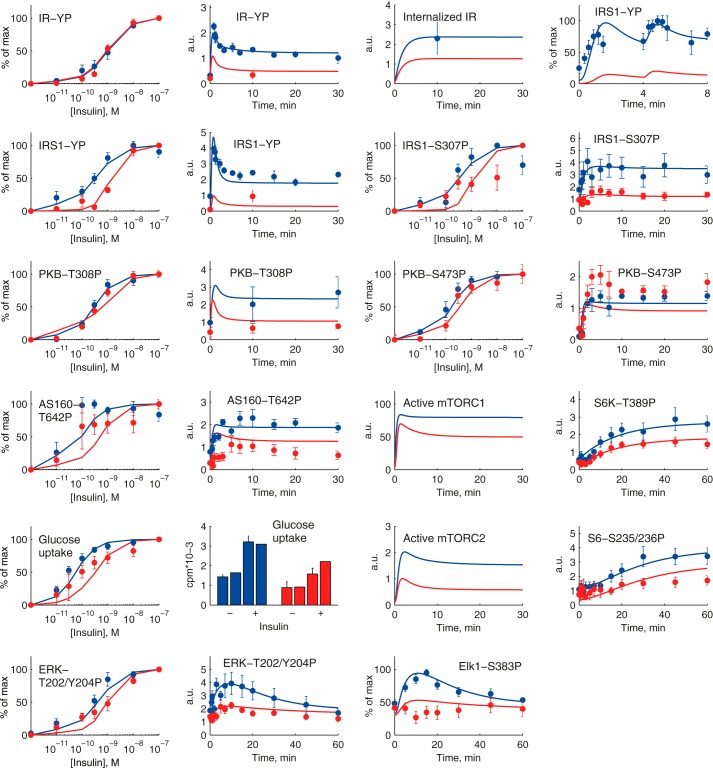
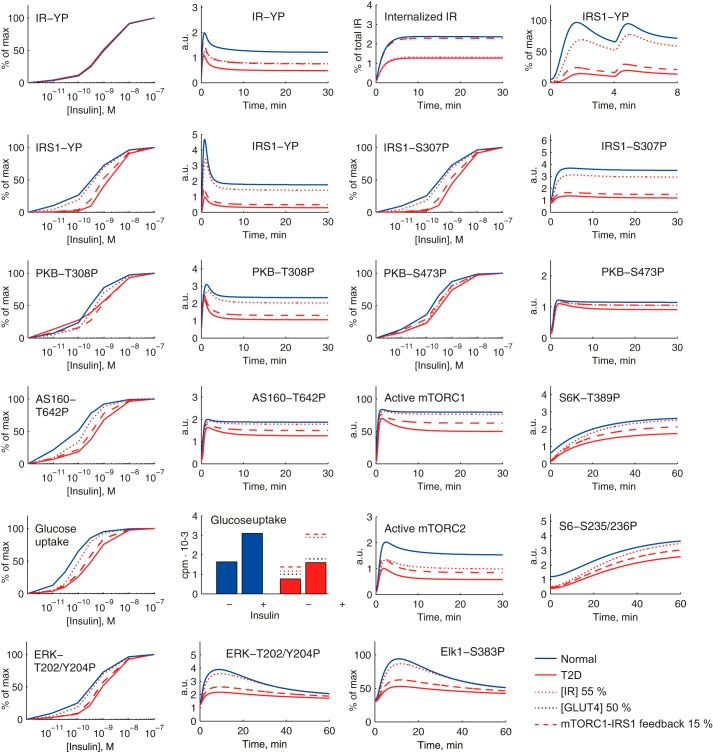
The response to insulin is impaired in type 2 diabetes. Much information is available about insulin signaling, but understanding of the cellular mechanisms causing impaired signaling and insulin resistance is hampered by fragmented data, mainly obtained from different cell lines and animals. We have collected quantitative and systems-wide dynamic data on insulin signaling in primary adipocytes and compared cells isolated from healthy and diabetic individuals. Mathematical modeling and experimental verification identified mechanisms of insulin control of the MAPKs ERK1/2. We found that in human adipocytes, insulin stimulates phosphorylation of the ribosomal protein S6 and hence protein synthesis about equally via ERK1/2 and mTORC1. Using mathematical modeling, we examined the signaling network as a whole and show that a single mechanism can explain the insulin resistance of type 2 diabetes throughout the network, involving signaling both through IRS1, PKB, and mTOR and via ERK1/2 to the nuclear transcription factor Elk1. The most important part of the insulin resistance mechanism is an attenuated feedback from the protein kinase mTORC1 to IRS1, which spreads signal attenuation to all parts of the insulin signaling network. Experimental inhibition of mTORC1 using rapamycin in adipocytes from non-diabetic individuals induced and thus confirmed the predicted network-wide insulin resistance.
Keywords: Adipocyte; Elk1; Extracellular Signal-regulated Kinase (ERK); Human; Insulin Resistance; MAPK; Mathematical Modeling; Protein Phosphorylation; Signal Transduction; Type 2 Diabetes.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Systems-wide Experimental and Modeling Analysis of Insulin Signaling through Forkhead Box Protein O1 (FOXO1) in Human Adipocytes, Normally and in Type 2 Diabetes.J Biol Chem. 2016 Jul 22;291(30):15806-19. doi: 10.1074/jbc.M116.715763. Epub 2016 May 20. J Biol Chem. 2016. PMID: 27226562 Free PMC article.
-
Insulin signaling in type 2 diabetes: experimental and modeling analyses reveal mechanisms of insulin resistance in human adipocytes.J Biol Chem. 2013 Apr 5;288(14):9867-9880. doi: 10.1074/jbc.M112.432062. Epub 2013 Feb 11. J Biol Chem. 2013. PMID: 23400783 Free PMC article.
-
Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes.Mol Med. 2010 Jul-Aug;16(7-8):235-46. doi: 10.2119/molmed.2010.00023. Epub 2010 Mar 26. Mol Med. 2010. PMID: 20386866 Free PMC article.
-
Metabolism and insulin signaling in common metabolic disorders and inherited insulin resistance.Dan Med J. 2014 Jul;61(7):B4890. Dan Med J. 2014. PMID: 25123125 Review.
-
The multifaceted role of mTORC1 in the control of lipid metabolism.EMBO Rep. 2013 Mar 1;14(3):242-51. doi: 10.1038/embor.2013.5. Epub 2012 Feb 12. EMBO Rep. 2013. PMID: 23399656 Free PMC article. Review.
Cited by
-
A comprehensive mechanistic model of adipocyte signaling with layers of confidence.NPJ Syst Biol Appl. 2023 Jun 7;9(1):24. doi: 10.1038/s41540-023-00282-9. NPJ Syst Biol Appl. 2023. PMID: 37286693 Free PMC article.
-
Insulin and β-adrenergic receptors mediate lipolytic and anti-lipolytic signalling that is not altered by type 2 diabetes in human adipocytes.Biochem J. 2019 Oct 15;476(19):2883-2908. doi: 10.1042/BCJ20190594. Biochem J. 2019. PMID: 31519735 Free PMC article.
-
A systems biology analysis of lipolysis and fatty acid release from adipocytes in vitro and from adipose tissue in vivo.PLoS One. 2021 Dec 31;16(12):e0261681. doi: 10.1371/journal.pone.0261681. eCollection 2021. PLoS One. 2021. PMID: 34972146 Free PMC article.
-
Current strategies and findings in clinically relevant post-translational modification-specific proteomics.Expert Rev Proteomics. 2015 Jun;12(3):235-53. doi: 10.1586/14789450.2015.1042867. Epub 2015 May 8. Expert Rev Proteomics. 2015. PMID: 25955281 Free PMC article. Review.
-
A multi-scale digital twin for adiposity-driven insulin resistance in humans: diet and drug effects.Diabetol Metab Syndr. 2023 Dec 4;15(1):250. doi: 10.1186/s13098-023-01223-6. Diabetol Metab Syndr. 2023. PMID: 38044443 Free PMC article.
References
-
- Carlson C. J., Koterski S., Sciotti R. J., Poccard G. B., Rondinone C. M. (2003) Enhanced basal activation of mitogen-activated protein kinases in adipocytes from type 2 diabetes. Potential role of p38 in the downregulation of GLUT4 expression. Diabetes 52, 634–641 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
