

Illumina human exome genotyping array clustering and quality control
- PMID: 25321409
- PMCID: PMC4441213
- DOI: 10.1038/nprot.2014.174
Illumina human exome genotyping array clustering and quality control
Abstract
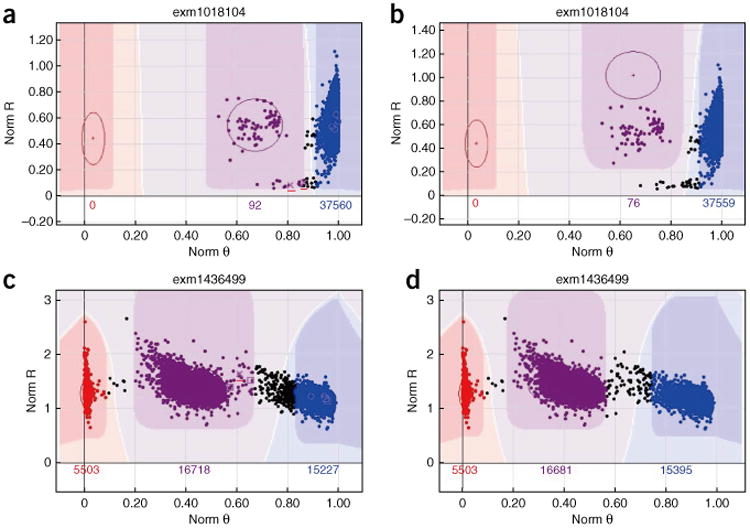
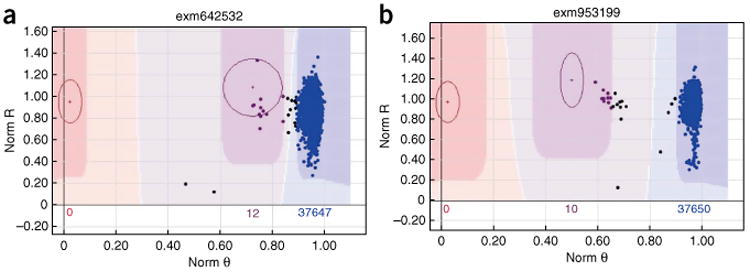
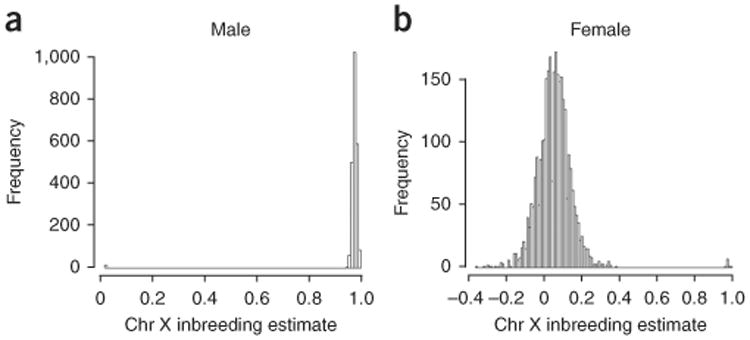
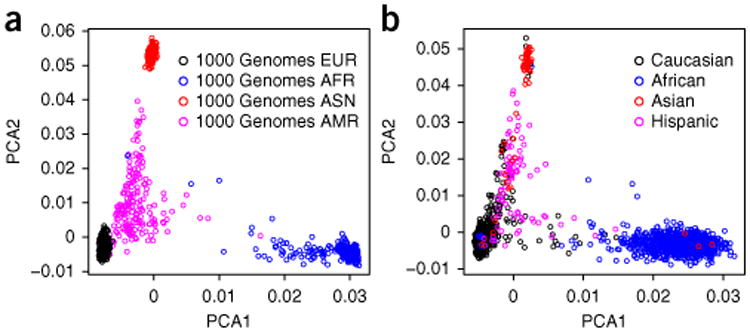
With the rise of high-throughput sequencing technology, traditional genotyping arrays are gradually being replaced by sequencing technology. Against this trend, Illumina has introduced an exome genotyping array that provides an alternative approach to sequencing, especially suited to large-scale genome-wide association studies (GWASs). The exome genotyping array targets the exome plus rare single-nucleotide polymorphisms (SNPs), a feature that makes it substantially more challenging to process than previous genotyping arrays that targeted common SNPs. Researchers have struggled to generate a reliable protocol for processing exome genotyping array data. The Vanderbilt Epidemiology Center, in cooperation with Vanderbilt Technologies for Advanced Genomics Analysis and Research Design (VANGARD), has developed a thorough exome chip-processing protocol. The protocol was developed during the processing of several large exome genotyping array-based studies, which included over 60,000 participants combined. The protocol described herein contains detailed clustering techniques and robust quality control procedures, and it can benefit future exome genotyping array-based GWASs.
Figures
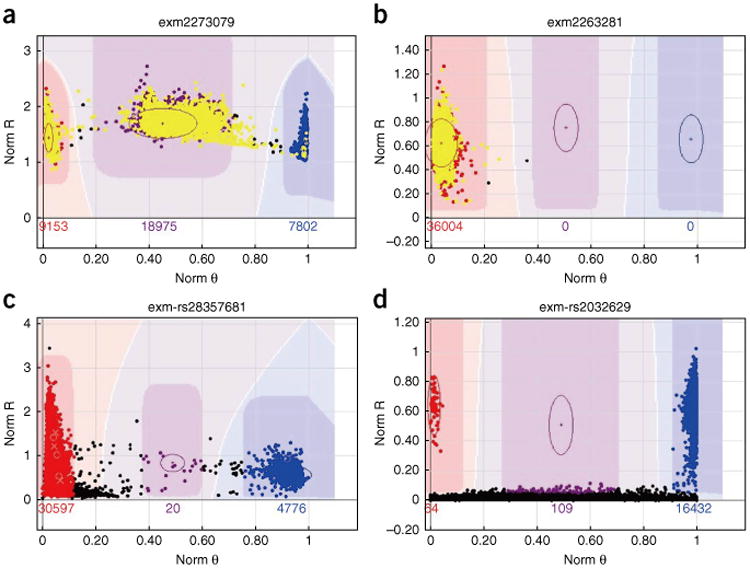
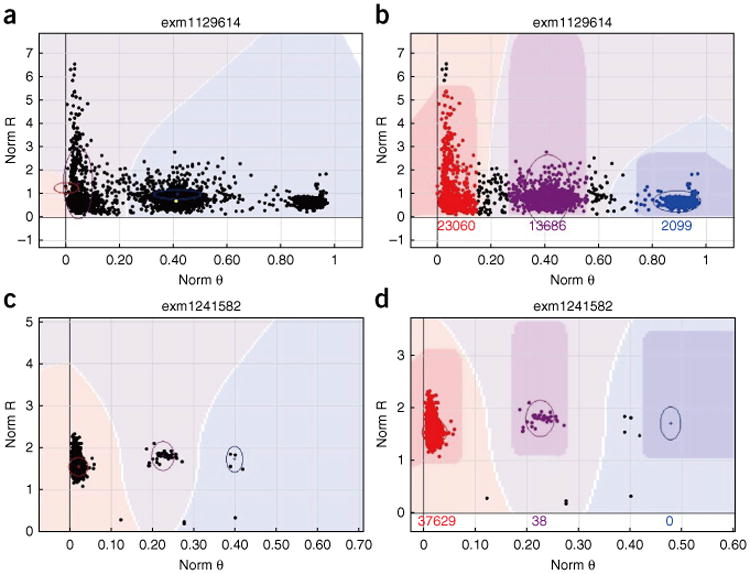
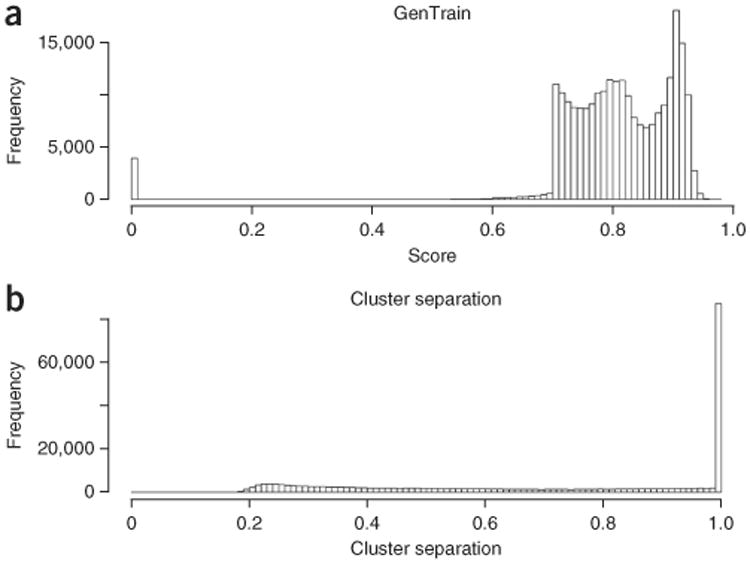
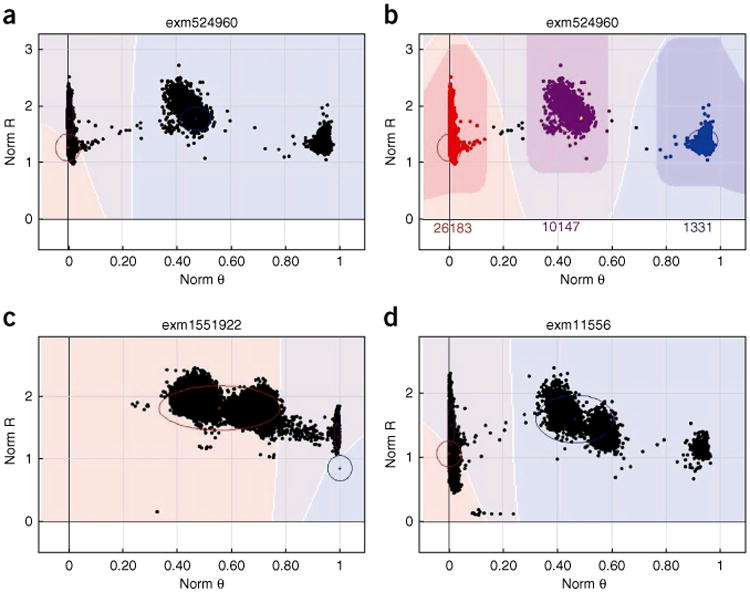
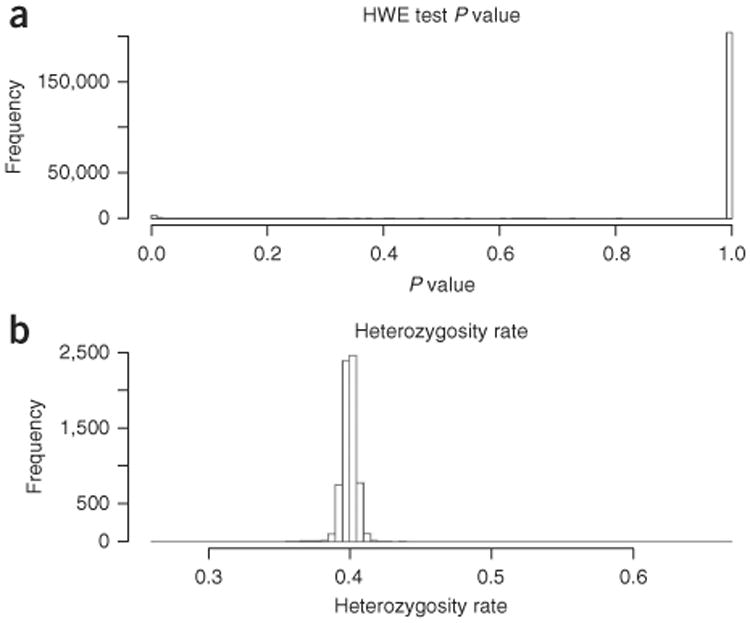
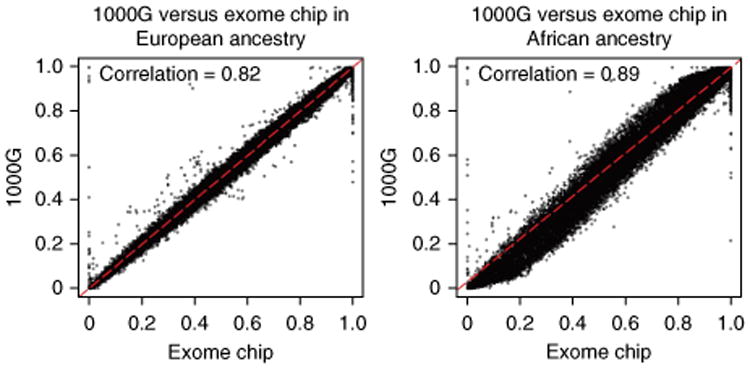
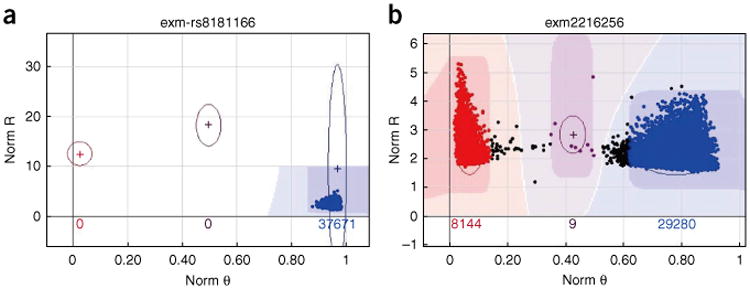
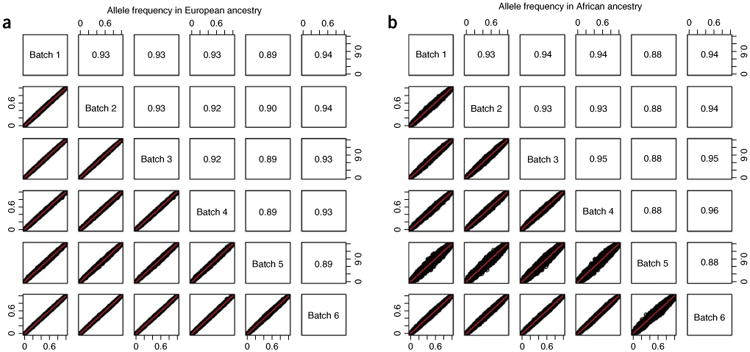
References
-
- Abecasis Lab. Exome Chip Design Wiki Site. http://genome.sph.umich.edu/wiki/Exome_Chip_Design.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
