

Whole-exome sequencing identifies homozygous GPR161 mutation in a family with pituitary stalk interruption syndrome
- PMID: 25322266
- PMCID: PMC4283017
- DOI: 10.1210/jc.2014-1984
Whole-exome sequencing identifies homozygous GPR161 mutation in a family with pituitary stalk interruption syndrome
Abstract
Context: Pituitary stalk interruption syndrome (PSIS) is a rare, congenital anomaly of the pituitary gland characterized by pituitary gland insufficiency, thin or discontinuous pituitary stalk, anterior pituitary hypoplasia, and ectopic positioning of the posterior pituitary gland (neurohypophysis). The clinical presentation of patients with PSIS varies from isolated growth hormone (GH) deficiency to combined pituitary insufficiency and accompanying extrapituitary findings. Mutations in HESX1, LHX4, OTX2, SOX3, and PROKR2 have been associated with PSIS in less than 5% of cases; thus, the underlying genetic etiology for the vast majority of cases remains to be determined.
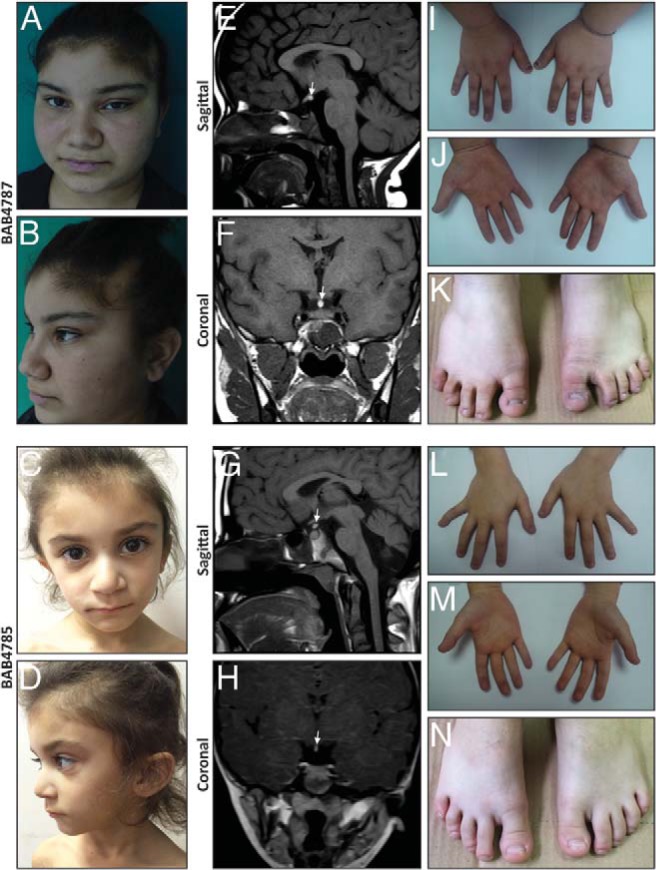
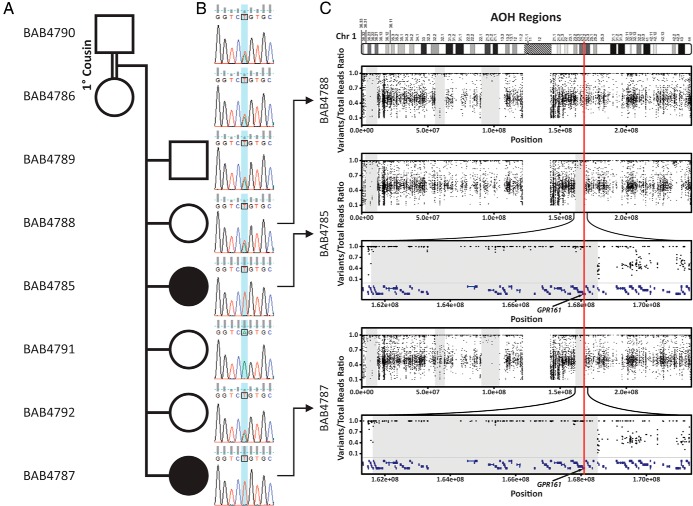
Objective: We applied whole-exome sequencing (WES) to a consanguineous family with two affected siblings who have pituitary gland insufficiency and radiographic findings of hypoplastic (thin) pituitary gland, empty sella, ectopic neurohypophysis, and interrupted pitiutary stalk-characteristic clinical diagnostic findings of PSIS.
Design and participants: WES was applied to two affected and one unaffected siblings.
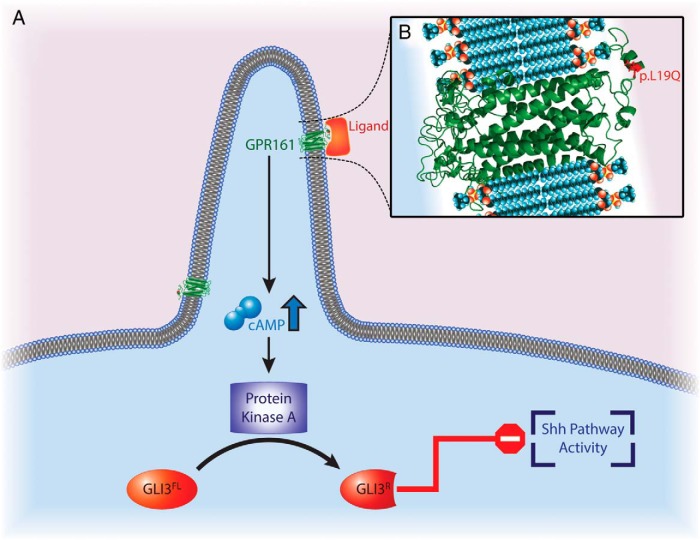
Results: WES of two affected and one unaffected sibling revealed a unique homozygous missense mutation in GPR161, which encodes the orphan G protein-coupled receptor 161, a protein responsible for transducing extracellular signals across the plasma membrane into the cell.
Conclusion: Mutations of GPR161 may be implicated as a potential novel cause of PSIS.
Figures
References
-
- Tatsi C, Sertedaki A, Voutetakis A, et al. Pituitary stalk interruption syndrome and isolated pituitary hypoplasia may be caused by mutations in holoprosencephaly-related genes. J Clin Endocrinol Metab. 2013;98:E779–784. - PubMed
-
- Pinto G, Netchine I, Sobrier ML, Brunelle F, Souberbielle JC, Brauner R. Pituitary stalk interruption syndrome: A clinical-biological-genetic assessment of its pathogenesis. J Clin Endocrinol Metab. 1997;82:3450–3454. - PubMed
-
- Reynaud R, Albarel F, Saveanu A, et al. Pituitary stalk interruption syndrome in 83 patients: Novel HESX1 mutation and severe hormonal prognosis in malformative forms. Eur J Endocrinol. 2011;164:457–465. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
